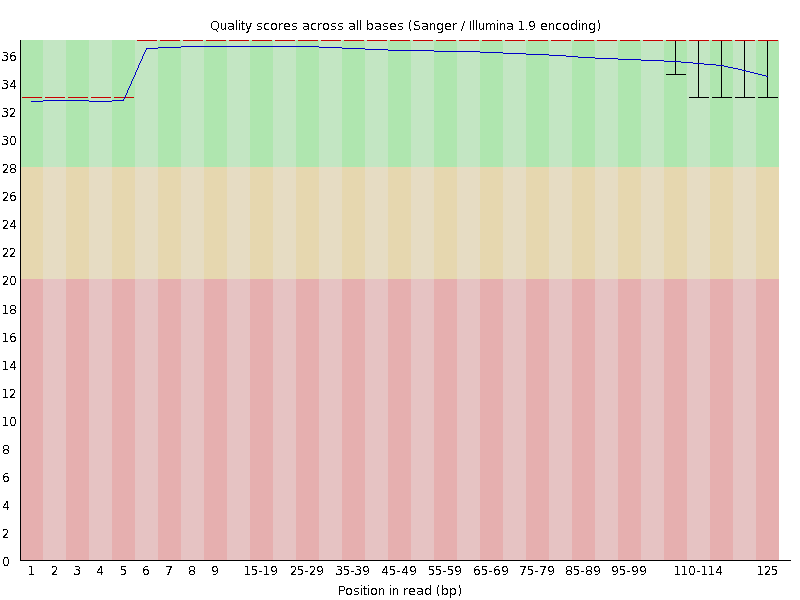
The first four cycles of data are used for cluster calling, and also for establishing metrics (e.g., signal thresholds) for base-calling. Base-calls for the first cycles rely on a standard set of parameters, but for subsequent cycles have been calibrated to the actual data (and are therefore more accurate).
Illumina also corrects the signal for phasing. Each cluster contains ~1000 copies, and imperfect chemistry means that some molecules are +1 or -1 relative to the actual cycle. Base-calling accuracy is improved by correcting the measurement (filtering the signal based on the preceding and subsequent cycles). Phase correction for cycle five is partially dependent upon the lower-quality preceding cycle, so it's quality is also lower (either that, or the algorithm doesn't use cycle four for phase correction). This is the same reason why quality of the last base is always significantly lower, since there's no subsequent cycle for phase correction.
为什么在read的质量值逐渐降低?
The really specific answer depends on what platform you're using but I'll go out on a (short) limb and guess its Illumina. If so, the drop-off is a phasing error.
With Illumina, DNA fragments are first bound to a flow cell. A well-prepared flow cell has even spacing between all DNA fragments. Before sequencing, the DNA fragments are amplified with a technique called bridge amplification, resulting in clusters of the same DNA molecule at each spot. Ideally no clusters overlap with each other (this is important for distinguishing clusters from each other). Illumina sequencers wash the flow cell with all 4 nucleotides and a blocker chemical so that only 1 base gets added to each molecule of DNA at a time. Different clusters may add different bases, but within a cluster it should always be the same.
This is how things work in a perfect world. In reality, a few molecules in each cluster will likely fail to add a nucleotide. So lets say we're on cycle 50 of 150. In this cycle 10 out of 1000 molecules fails to add a new nucleotide (an A). Next cycle (cycle 51) when the 990 other molecules add a G, the 10 that failed last cycle will add the A instead. From now on they will be at least 1 cycle behind the rest, polluting the light signal that the sequencer's camera has to read. in cycle 52, some more sequences fall behind from the main group, and some from the group that was already behind fall even further behind. You can see that by cycle 150, several percent of the molecules may well be out-of-sync with the cycle number and by the end of the sequencing run, the last N bases will have a less pure light signal than the first. This is the information Illumina sequencers use to calculate quality scores. New chemistries are largely intended to minimize this phasing problem, increasing the length of reads before quality begins to drop.
Reference:
WHY are the first few bases of Illumina HiSeq reads of lower quality?
http://seqanswers.com/forums/showthread.php?t=61936
Why does base quality of reads generally decreases at the end of the read?
https://www.biostars.org/p/177027/