Protein&Cell综述|解析宿主基因组和微生物的复杂互作(王军组作品,万字全文翻译)...
本文转载自“热心肠先生”,己获授权。

2018年5月,Protein & Cell 杂志特别为中国肠道大会推出了主题为“Microbiota and Human Health”的专刊,邀请于君、傅静远、马永慧、王军、张发明、赵方庆、陈峰、王则能、聂勇战等9位教授领衔发表了9篇综述和评论文章,并由刘志华教授撰写卷首语。
Protein & Cell 杂志是开放获取期刊,所有文章的pdf版本都是免费下载的,大家可以点击访问 http://www.protein-cell.org/archive/index-2018-5.html 获得该期专刊的英文版原文。
今天我们发出来自中国科学院微生物研究所王军研究员、朱宝利研究员和团队的综述的翻译稿,我们特别感谢英国帝国理工学院在读博士生陈绮翎同学为翻译本文所付出的辛勤努力。

5月12日,王军研究员曾在国家会议中心的中国肠道大会上系统讲解本综述,大会的付费参会者和演讲嘉宾可访问 http://www.mr-gut.cn/videos/show/9092212061 观看演讲讲解视频。
希望能帮到你涨知识,不用谢,我们都是热心肠!
解析宿主基因组中基因和微生物的复杂互作
Of genes and microbes: solving the intricacies in host genomes
作者:Jun Wang(王军), Liang Chen, Na Zhao, Xizhan Xu, Yakun Xu, Baoli Zhu(朱宝利)
通讯作者:王军,[email protected];朱宝利,[email protected]
关键词:肠道菌群,宿主遗传学,数量遗传学,基因-微生物组互作,共生总基因组
译者:陈绮翎同学(英国帝国理工学院在读博士生)
摘要
微生物组研究是生物医学研究中一个迅速发展的领域,我们已经见证了其在研究生理学、代谢组学和免疫学中的潜力,见证了其在宿主健康和疾病中扮演的重要角色,也见证了其在疾病预防、干预和治疗中的巨大作用。
但是,这个领域仍有许多基本问题亟需解决,包括导致不同个体之间和不同时间点之间微生物多样性变化的因素。
塑造微生物组的有先天和后天因素,宿主基因会部分塑造微生物组,而环境因素则会改变微生物组发展的原始过程。
在这篇综述中,我们聚焦于宿主遗传等先天因素,并归纳总结近期关于基因组对微生物群落的组成和功能产生重要影响的研究,这里的基因组尤指人类和小鼠的基因组,微生物群落主要指位于肠道但也包括皮肤表面等部位。
我们旨在概述用于研究宿主基因变异和微生物间复杂关系的不同方法,以及这些方法背后的哲学和方法论,我们也旨在总结探索过程中的一些关键性发现和既有局限性。
在未来的研究中,一定会出现更多证据和成果,而持续积累的知识会让我们更深入地理解什么是“共生总基因组(Hologenome)”,也就是宿主和微生物组之间有序组织、紧密互作的基因组。
引言
有人体自身细胞数约1.3倍的细菌定植在我们身体里 (Sender等,2016),它们大部分存在于胃肠 (GI) 道中 (Qin等,2010;Zhu等,2010),很难想象我们的基因组不会用一些特定的基因来应对潜在的威胁,并与我们的微生物组协同产生有利作用。
事实上,现在在人类、其他动物 (Kurilshikov等,2017) 甚至植物 (Lundberg等,2012) 中有很多基因-微生物组互作的迹象,它们中很大一部分在广泛使用二代测序技术之前就已经被鉴定出来。
那些基因作用于免疫系统(Hooper等,2012)是合乎情理的:病原体(其中一大部分是细菌)是塑造人类基因组进化、促进依赖于免疫系统来抵抗病原体存活下来的最重要推动力量之一 (Kau等,2011)。
在动植物的自然种群中,流行病的出现会不断地消灭局部种群(导致某一地区特定物种的消失)或全部种群(导致大灭绝)。但是,一旦在这些流行病中出现了幸存者,我们通常可以在它们的基因组中找到解释,比如和免疫相关的基因出现自然变异使得某一特定种群出现高抗性并存活下来 (Brinkworth和Pechenkina,2013)。
在下一代中,那些等位基因(特定基因的一个变种)出现的频率通常会增加,导致种群基因发生变化 (Prugnolle等,2005)。事实上,更多病原体并非和能导致流行病的病原体一样有杀伤力,它们导致的致死感染会较少且只会危害少数宿主的健康;然而这些病原体仍然能够导致等位基因频率的变化 (Barreiro和Quintana-Murci, 2010)。
当然,病原体也涉及宿主和病原体之间的生存竞争;但相反,没有任何特定等位基因会是完美的解决方案,在不同的时间段增多不同的等位基因的是有利选择的结果 (Novembre和Han, 2012)。
拜历史和医学记录所赐,我们清楚地知道人类过去和现在面对的最大威胁,我们还可以从我们的基因组中看到这些威胁的影子 (Barreiro等,2008)。
黑死病曾经一次性杀死欧洲三分之一的人口,其影响甚至可以从现代欧洲人口中看到,包括一些在《病者生存》一书中被归纳的出人意料的后果 (Moalem和Prince, 2008)。尽管每年几乎只在很小范围存在,但它依然在发生。
目前,结核病 (TB) 在全球感染数百万人,主要是在不发达地区 (世界卫生组织,2016); 在抗生素被发明之前,这些感染范围更广。Jostins等(2012)惊讶地发现,我们认为会导致炎症性肠病 (IBD) (主要包括克罗恩病、溃疡性结肠炎以及自身免疫病,其影响一小部分欧洲人口) 的基因其实是TB选择后的结果。
通过尚未被人知晓的机制,那些基因或降低免疫系统对感染的敏感性,或利用细菌阻碍识别位点,以此加强我们免疫系统对TB的抵抗力;因此,这些基因仍处于被病原体选择的阶段,而且其频率会在种群中不断发生变化(Jostins等,2012)。
我们现在知道上百种复发性细菌感染,霍乱和细菌性脑膜炎就是其中的两种,这些复发性病原细菌很多已经在我们的基因组中留下了记号 (Gupta, 2016) (图1)。
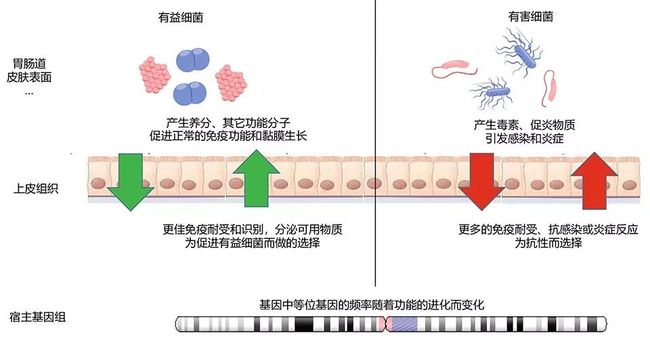
图1. 在不同上皮细胞交界处宿主基因-微生物组相互作用的简化图。
胃肠道的黏膜层、呼吸道、皮肤表面和生殖道表面是宿主-微生物相互作用的主要场所。
那些我们认为有益的微生物通常生产营养物质、必要的功能分子并维持免疫系统的正常功能;因此宿主基因的主要目的是确保它们的免疫耐受性,并通过分泌黏液等物质促进它们的生长。
当有害细菌通常生产毒素、促炎分子并导致感染时,宿主基因必须将它们从正常的群落中移除,并抵御炎症和感染。
然而,故事并不总是有关于“坏”细菌。
尤其是在最近十年,我们开始了解人类和动物的胃肠道中 (Spor等,2011) 、皮肤上 (Grice和Segre,2011) 、口腔内 (De Whirst等,2010)以及生殖 (Ravel等,2011) 和呼吸系统 (Dickson和Huffnagle,2015) 中复杂的微生物群落的组成和功能。
另外,我们已经意识到正常的微生物组在我们自身的健康中起到重要作用 (图1)。我们依赖肠道微生物组去消化食物并将大分子物质代谢成小分子物质,从而肠道可以轻而易举地吸收它们 (Kau等,2011)。
肠道微生物组制造大量的其它产物,包括维生素、血清素和许多调控宿主体内不同系统的功能分子 (Kau等,2011;Kostic等,2014);因此,肠-脑轴 (Foster和Neufeld, 2014),肠-肝轴 (Ray, 2017) 和肠-肺轴 (Budden等,2017) 的概念得以出现,经过检验后被广为接受。
微生物组促进婴儿免疫系统的早期发育,维持成年人正常的免疫功能;同时,许多和免疫相关的疾病主要是由微生物组的紊乱造成的 (Kamada等,2013)。
在本领域被广泛认可的“共生总基因组(Hologenome)”概念可以被理解为,在基因组和宏基因组水平上,全面性囊括宿主及其微生物组在针对自然选择和进化时,组成功能性实体和基础所需的全部互作、合作和互选(Zilber-Rosenberg和Rosenberg,2008)。
间接证据
尽管我们归纳在这里的研究并不严格按照时间顺序排列,但我们还是希望能够于让读者沿着历史的路径去探究基因-微生物组互作。
比如,我们已经知道许多基因有可能对于维持宿主抵抗病原体的能力 (主要组织相容性复合体,MHC) (Neefjes等,2011)和感知微生物的能力 (Toll样受体,比如TLR,能够感知许多由微生物产生的分子) (Kieser和Kagan,2017) 极为重要,也有一些基因可能会涉及其它重要的会导致疾病的过程。
但是,这些主要是基于自然敲除模型(如一个会导致某一特定基因功能丢失的突变)得到的结论。我们通常研究的是处于不健康状态的小鼠或人类,因为宿主-微生物互作中的关键基因已经不再起作用了,因此可以表明基因功能图谱的极端情况。
直到最近,更宏观地去观察整个基因组变化的研究才出现,尤其是中性或几乎中性的等位基因(那些不会导致类似功能丢失一样危害的基因)与它们和微生物组作用的关系的研究 (比如,Hov等,2015;Wang等,2016;Bonder等,2016;Turpin等,2016;Goodrich等,2016)。
Ochman研究组在Plos Biology上发表了一项包括人类在内的,关于人科——灵长类动物的研究,结果显示,微生物组的歧化和可以高度体现宿主系统发育的亚基因组——线粒体基因组的系统发育完美对齐(全等)(Ochman等,2010)。这项研究之后还有几项其它的深度研究 (Moeller等,2014;Nishida和Ochman,2017)。
这项研究利用Unifrac距离来估计微生物组的歧化 (Lozupone等,2011), Unifrac距离是细菌关系的总体系统进化程度,它考虑了细菌种类的丰度以及它们在系统发育体系中的位置。
然后,微生物组的总体不同也被集群形成“系统发育树”来显示它们的相对相似性,而其和宿主系统发育的全等表明,微生物组的不同确实能够被宿主基因的不同来加以塑造。
但是,必须注意的是,这里的证据并非没有潜在的混淆因素,尤其是考虑到微生物组和它的宿主在进化和歧化过程中的变化,以及不同宿主的种间饮食差异和地域隔离等 (Bodenstein & Theis, 2015)。
这已经在其它的比较研究中被注意到,研究者试图将基因雷同的标记从环境类似物中分离出来,尤其是饮食 (Ley等,2008)。
尽管如此,这些研究还是使我们开始明白,在共生总基因组中,宿主间的差异对肠道微生物间的变化有重大影响,至少是在不同物种间的差异。
Ley研究组利用英国双胞胎生物样本库 (the UK Twins biobank) 进行了另一项里程碑式的研究 (Goodrich等,2014)。总体思路相当直接明了:通过比较基因完全相同(同卵)或不相同但相关(异卵)的双胞胎,人们可以迅速确定是否有一些性状(在这里是微生物组)与宿主基因具有相关性。
研究假设是,在双胞胎中,环境带来的差异是最小的,或者至少在同卵和异卵双胞胎之间不会是极度不同的。且在前者(同卵双胞胎)中,当某一性状相较于后者(异卵双胞胎)更相似时,这一定是由于基因相似性造成的。
通过使用16S rDNA和宏基因组学分析进行一系列的后续研究,他们确实在英国双胞胎的人体肠道微生物组中发现了这个现象 (Goodrich等,2016;Xie等,2016)。
几个独特的细菌也显示出相当高的遗传力,因为相同的遗传特点,所以被定义为是一个性状的相似性,包括和身高体重指数 (BMI) 负相关的Christenalleaceae科细菌。小鼠模型也显示这科细菌确实有减少肥胖的作用。
但让人失望的是,进一步分析定位与该科细菌相关的宿主基因位点,并未得到一个确定的基因,这可能是由于双胞胎样本数太少的原因。全基因组关联分析 (GWAS)通常需要相对无关的个体,而对于双胞胎,有效样本数被减半且达不到1000。
Org等(2015)在113种不同品系的小鼠中进行了相似性分析,相比不同品系小鼠个体之间的微生物组差异,同一品系小鼠不同个体之间的微生物组更相似。他们估计了微生物组的遗传力,把那些不同品系小鼠之间的关联和谱系算在内,总结得出宿主基因变异能够解释肠道菌群的大量变异。
直接证据:可控基因研究
解决混淆因素问题
相对于宿主基因相对稳定的特点,微生物组更倾向于是一个动态系统,它有自己的自然波动,并且很容易被许多不同的环境因素影响 (Hall等,2017);因此,同一个体在不同时间点的微生物组可能会非常不同。
此外,当我们关注横断面研究时,大部分的大规模研究会有相对应的规模局限性,我们是在检测不同个体之间微生物组的瞬时图谱,并伴随很强的随机性和噪音 (Walter和Ley,2011)。
但是,这个和生物学的很多领域都类似:我们依赖足够强的、能够被适当的探测方法识别的生物学信号,且在此框架下,也包括统计方法学。另外,我们依赖足够大的样本量,去把具有统计学意义的信号从其它信号中分辨出来。
尽管如此,阐明最重要的混淆因素对任何基因学研究都十分重要,如果不考虑的话,会导致I类(假阳性,错误的基因位点显示具有意义)和II类错误(假阴性,真正的基因位点被噪音覆盖)。
在小鼠模型中,我们能够控制那些混淆因素来最小化它们,而在人类中,这就需要对混淆因素进行系统性分析。
大量的研究纳入了人体测量结果,包括年龄(Yatsunenko等,2012)、BMI(Dominianni等,2015)、腰围和饮食习惯(David等,2014;Dominianni等,2015)以及其它生活习惯等(Yassour等,2016)。
2016年,在Science杂志的特刊中同时出现了两篇报道,这两篇报道分别有关于比利时佛拉芒人(Flemish Gut Flora Project)(Falcony等,2016)和荷兰北部人(LifeLines-DEEP cohort)(Zhernakova等,2016),研究人员以人群为基础,分析影响微生物组的多样性的混淆因素。
在这项研究中,上百个不同的测量数据被测出、过滤,然后分别按照它们对以下指标的贡献定级:肠道微生物组总体差异性(β-多样性),类群丰度——也被称为α-多样性的总体特性,以及功能能力。
许多被研究的因素(包括性别、BMI、血液化学等),部分是由基因决定的,也因此表明基因会参与微生物组的演化。类似于年龄的其它因素,是绝对不由基因决定的,但是当这些因素是主要的参与要素时,基因学研究必须对此进行解释。
现在,这可能听起来很奇怪,当我们研究基因学时,我们也需要对混淆因素进行控制。
基本原理如下:在使用杂交种群(数量性状位点,QTL)或自然种群(全基因组关联分析,GWAS)进行定量的基因学研究中,我们意识到一个性状的相似性可能是因为总体的关联性。
比如,同父同母的小鼠有着十分相近的成长环境,并分享来自同一母系传播的微生物组(Benson等,2010;Wang等,2015)。有关联的人类个体间,因为同样的原因可能也具有相似的微生物组 (Goodrich, 2016)。
相反地,如果我们研究的种群没有很好的混合,存在亚种群,并因此提供一个特定的人群结构,那么我们发现的在不同个体间的性状可能不是因为几个基因的作用,而是进化、分离、漂移等等的长期历史性累积的作用 (Yatsunenko等,2012)。
在QTL研究和GWAS分析中,说明亲属关系并完全确定是否存在特定的人口结构很有必要。
通常来说,除了相关的特定个体,其它的都会在GWAS中被移除,并且许多人试着尽可能的同质化被研究的人口。但是,也有数学方法可以将亲属关系算在内,或者通过基因主成分将人口结构算在内(Kang等,2008;Price等,2010)。
我们快速讨论下用来阐明混淆因素的方法,但不会深入太多技术细节。
当我们对大部分重要的混淆因素进行单变量特征研究(如丰度或类群丰度)时,我们使用线性模型/广义线性模型来移除它们的“作用”,并将剩余的部分留给基因分析。
这种方法相对直接但有时候不能被考虑周全,因为许多微生物组对一个因素的响应并非线性 (Lahti等,2014);但是,其它非参数因素也未必表现更佳,并可能会在其残余部分中起误导作用。
对于微生物组的整体差异beta多样性),我们也可以通过使用限制性主成分分析(PCoA)去除特定因素的混淆作用,然后分析其残余部分(也是一个距离矩阵)(Ruhlemann等,2017)。我们很少看到PCoA被使用,主要是因为至今很少有人研究beta-多样性和宿主基因组的关联性,且这个领域仍处于初级阶段。
候选基因方法
基于历史性、医学和政策原因,IBD仍是许多微生物组研究的焦点。在欧洲,这是一种普遍的胃肠道慢性炎症,其发生率大约是1%,而且主要出现在犹太人后裔中(Hanauer, 2006)。
一系列基因学研究显示,在其它风险因素中,IBD患者一长串的潜在基因风险因素包括NOD2, CARD9, ATG16L1, IRGM和FUT2(Xavier和Podolsky, 2007)。
因为微生物组因素在IBD中占了很高的比例,许多风险基因都被用来检测是否对微生物组有影响 (Kostic等,2014)。
许多IBD基因风险因素确实和Ruseburia菌属的减少显著相关,该菌属在把乙酸转化为丁酸的过程中起到重要作用,相比健康对照组,Ruseburia菌属在IBD患者体内减少(Morgan等,2012)。
我们已经归纳出假定的对微生物组有作用的基因,并依次在人(自然的基因变异)或小鼠(敲除模型)中进行了检测。我们可以看到,大部分研究仍集中在IBD上。当然,这个列表不完整的,但包括了我们知道的大部分重要基因(表1)。
表1. 在宿主基因-微生物互作中利用候选基因方法研究的例子。
我们进行了关于宿主基因、微生物组和疾病的文献搜索,并列出了大部分重要的例子,这些例子中关于微生物组变化的假定驱动型研究或是基于人类(自然基因变异)或是基于小鼠(敲除模型)。
我们列出了观察到的变化和研究背景(疾病类型),主要集中在IBD上。
基因名称 |
和基因变化相关的特征 |
研究背景 |
参考文献 |
人类 |
|||
IL13/CD14 |
与剖腹产过程及产前接触抗生素相互作用,影响皮肤微生物组 |
过敏性皮炎 |
Lee等(2014) |
FUT2 |
呼吸道微生物组(绿脓杆菌,Pseudomonas aeruginosa) |
支气管扩张 |
Taylor等(2017) |
IL6 |
幽门螺旋杆菌(Helicobacter pylori) |
血脂异常 |
Pohjanen等(2016) |
ATG16L1 |
梭杆菌科 (Fusobacteriaceae),拟杆菌科 (Bacteroidaceae),毛螺菌科 (Lachnospiraceae),肠杆菌科 (Enterobacteriaceae),大肠杆菌 (E. coli) |
IBD |
Sadaghian Sadabad等(2015) |
CARD9 |
肠道微生物组组成 |
IBD |
Lamas等(2016) |
FUT2 |
肠道微生物组组成、多样性和机构 |
IBD |
Rausch等(2011a, b) |
NLRP12 |
肠道微生物多样性 |
IBD |
Chen等(2017a, b) |
NOD2 |
肠道微生物组成 |
IBD |
De Bruyn等(2017) |
SLC39A8 |
肠道微生物组成 |
IBD |
Li等(2016) |
TNFSF15 |
普雷沃菌属 |
IBD |
Nakagome等(2017) |
SI |
Blautia, 摇摆杆属 (Oscillibacter), 瘤胃球菌属 (Ruminococcus)和未分类的肠杆菌科 |
IBS |
Thingholm等(2018) |
IFN-I |
和微生物相关的色氨酸代谢 |
多发性硬化 |
Rothhammer等(2016) |
DEFB-CN |
鼻咽癌细菌定植模式 |
中耳炎 |
Jones等(2014) |
A2ML1 |
中耳微生物组 |
中耳炎 |
Santos-Cortez等(2016) |
C4B |
肠道微生物组组成 |
儿科炎症性肠病 |
Nissila等(2017) |
CARD15 |
克罗恩病人体内牙周菌群 |
牙周炎 |
Stein等(2010) |
ELANE |
龈下菌群 |
牙周炎 |
Ye等(2011) |
小鼠 |
|||
Myd88 |
多样性,分段丝状细菌 |
抗菌信号 |
Larsson等(2012) |
Vdr |
乳杆菌属 (Lactobacillus),梭菌属 (Clostridium),拟杆菌属 (Bacteroides),Alistipes, Odoribacter, Eggerthella |
胆汁酸代谢 |
Jin等,(2015) |
Tnf |
肠道微生物组组成 |
结肠炎 |
Kozik等,(2017) |
Can |
大肠杆菌 |
大肠直肠癌 |
Peuker等,(2016) |
Lcn2 |
Alistipes |
大肠直肠癌 |
Moschen 等,(2016) |
Ifnar1 |
肠道微生物组组成 |
IBD |
Tschurtschenthaler等,(2014) |
II10/Tlr4 |
肠道微生物组组成 |
IBD |
Ward等,(2016) |
II2 |
大肠杆菌Nissle株,普通拟杆菌 (B. vulgatus) 和大肠杆菌mpk株/普通拟杆菌 |
IBD |
Bohn等,(2006) |
NIrp12 |
肠道微生物组组成 |
IBD |
Chen等,(2017a, b) |
Sirt1 |
肠道微生物组组成 |
IBD,大肠直肠癌 |
Lo Sasso等,(2014) |
Muc2 |
肠道微生物组组成 |
回肠稳态 |
Sovran等,(2015) |
Mhc |
肠道微生物组组成 |
免疫学 |
Kubinak等,(2015a, b) |
B4galnt2 |
肠道微生物组组成和沙门氏菌敏感性 |
炎症 |
Rausch等,(2015) |
TREM-1 |
肠道微生物组全面失调 |
炎症 |
Kokten等(2018) |
Nod2 |
高脂饮食下的肠道微生物群 |
肥胖 |
Rodriguez-Nunez等(2017) |
Fut2 |
肠道微生物组的多代动态学 |
肠道感染的敏感性 |
Rausch等(2017) |
有趣的是,在这些基因中有两个基因是表面聚糖的决定因素,它们是宿主-微生物互作的起始连接位点/分子。
第一,FUT2基因编码岩藻糖转移酶-2,这个转移酶涉及到在胃肠道黏膜和分泌物中发现的ABO血型抗原的表达。现发现有两个特定的基因型,一个是功能性分泌基因,一个是功能丢失导致无法分泌的变异基因 (McGovern等,2010)。
近期的研究显示,FUT2分泌物状态 (由基因型定义) 对肠道菌群有显著影响 (Rausch等,2011a, b);因此,Blautia菌属在A分泌组中比在非A分泌分组中少,并且这个减少伴随着理研菌科 (Rikenellaceae), 消化链球菌科 (Peptostreptococcaceae), 梭菌目 (Clostridiales) 和苏黎士杆属 (Turicibacter) 丰度的升高 (Gampa等,2017)。
有趣的是,小鼠基因B4galnt2(编码糖基转移酶β-1,4-N-乙酰半乳糖胺转移酶2)在确定肠道黏膜的糖组成方面有类似的功能,而且当我们考虑表达模式的时候它具有组织特异性。其在肠道中是否表达和小鼠模型中变化的细菌群落组成紧密相关 (Staubach等,2012)。
B4galnt2的肠道表达改变了肠道微生物组,并因此增加了肠道沙门氏菌(Salmonella typhimurium)对上皮组织的侵染,其潜在的机理可能是通过增加肠道炎症性细胞因子以及浸润免疫细胞。另外,B4galnt2在小鼠种群中有一个有趣的选择特征,我们将在最后讨论。
另一系列例子是负责感知微生物和引发下游细胞信号通路的基因。它们通常是先天免疫系统的组成部分。
比如,外源性微生物能够被模式识别受体 (PRR)识别,包括但不限于Toll样受体 (TLR) 和NOD-样受体 (Kieser和Kagan, 2017), 并且被MyD88基因编码的MyD88蛋白能够作为转换器调控信号转导途径。
对这些基因的研究已有应用了基因敲除的小鼠模型,并且现已发现这些基因对肠道微生物组的作用。
另外,我们发现MyD88信号对I型糖尿病(T1D)的形成至关重要,但是T1D在小鼠中的发病率会因为小鼠受到微生物刺激而降低,诸如分支杆菌属或不同的微生物产物,这些结果说明,特定基因之间的互作对于生命体早期免疫系统的健康发育很必要 (Wen等,2008;Kostic等,2015)。
但是,至今最耐人寻味的案例是MHC位点,不管是用候选基因方法还是最新的GWAS 方法(之后会讨论),人们始终无法找出其与肠道微生物组成的重要关系。
迄今为止,规模最大的研究是在挪威开展的一项利用骨髓标记来区别不同的MHC等位基因的研究,并且搜集到的微生物组没有显著的差别 (Hov等,2015)。
但是,在小鼠模型中就得出了不同的结论,这些信号非常突出 (Kubinak等,2015a, b) 。
这强调了在研究人类基因组学时很难确定其对微生物组的影响,特定基因差异的影响可能非常小(且在之后的研究中得到了验证),而且可能被环境差异覆盖掉了。在小鼠模型中,那些因素更容易控制。
要补充说明的是,我们须承认我们没有列出候选基因方法的全部基因列表,我们仅仅涉及了一些经典基因(表1)。
但是,基本原理是相同的,而且我们确实期待看到更大规模的此类研究,并且每个研究都能涉及对基因-微生物组相互作用机理的更深入探索。
数量遗传学
当我们试着去探究微生物组和宿主的全部基因组而非单个基因时,定量研究基因组学的工具就派上用场了。
数量遗传学主要缘起于植物和动物育种科学,其致力于寻找隐藏在被研究物种的重要生物学性状(表型)后的基因和基因位点,向因果、机制研究和实际应用(比如,改善作物或动物产品)提供基础理论支持(McCarthy等,2008)。
两个被广泛使用的方法:QTL和GWAS (图2)。
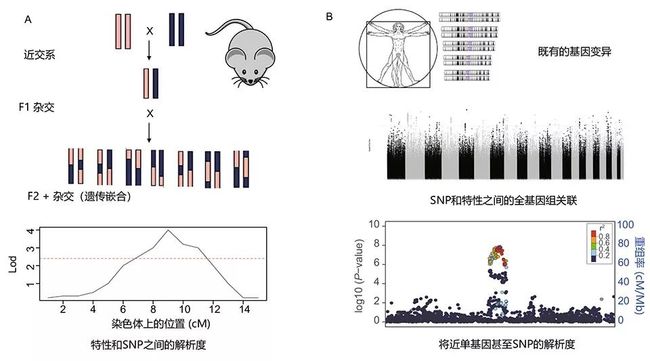
图2. 定量特征位点(QTL, 小图1)和全基因组关联分析(GWAS,小图2)的原理概述图。
二者均关于基因变化,但是源于或刻意杂交(QTL)或自然出现且尚存的(GWAS)不同过程,并且连锁区域因重组数目而具有不同大小。
QTL分析使用关联度测试对SNP进行检测,并将其插入一个区域,而GWAS对每一个SNP都进行检测,曼哈顿散点图中的一个“峰”代表一个可能和特征相关的单体型。
在这两个案例中,P值通常被转换为-log(P)来显示重要程度,对于QTL的全基因组的重要性则通常由置换检验决定。对于GWAS,通常被接受的该值为1E-08到5E-08。
许多人会说,严格地来讲,这两者不能同时存在,因为前者应用于定量性状,比如动物高度或作物产量,主要是利用提前设计好的杂合体作为研究群组,解析度和基因重组的产生成比例;而后者主要应用于人类,人类数千代的人口自然发展史导致了无数基因重组的出现。
但是,这并没有达到单个基因的水平。相反,人类基因组仍然存在大片连锁的基因,并且几乎还没有发生过重组。
与之相对的是,连锁不平衡 (LD) 也作为结果出现,这会使得关联性解析度出现在基因水平或甚至在SNP水平;许多案例中,被研究的特征是二元的,尤其是关于疾病。
但是,我们想要强调,从数学的角度来看,这两种方法在本质上是一样的。
数量遗传学研究意在寻找基因变异之间的显著关联性,不管是单个SNP之间还是一大片染色体区域之间。这两种都考虑了LD信息,以及特定表型变化和只影响相关测试模型的不同类型的特性。
二元特征需要逻辑回归,其关联性的结果通常被表示为优势比 (odds ratio,OR):相比一个特性的基础频率,一个特定的SNP能够通过OR的倍数来改变该特性的频率。因此,如果OR高于1,则频率增加,反之亦反。
连续分散的特性需要线性或类似的回归,而SNP/单体型区块的“作用”是由所使用的模型决定的β-值或Z-分值。这意味着和某个特定SNP/单体型区块相关的平均值,来源于通过方差测量得到的总体平均值 (Hirschhorn和Daly,2005)。
迄今为止,我们知道有6项使用杂交的QTL分析研究(图3),这些分析以小鼠为模式生物,以微生物为目标特性。其中4项QTLs以肠道微生物组为目标特性,剩余的2项关注皮肤菌群。
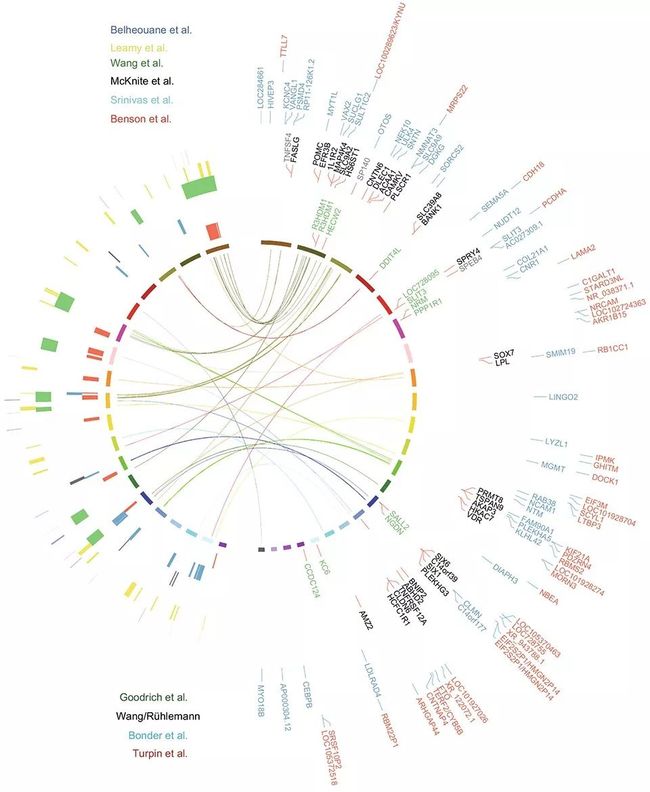
图3. 迄今为止小鼠微生物组QTL和人类GWAS的概述图。
左半部分显示了小鼠中的6项QTLs分析,用不同的颜色表示,置信区间被标记在小鼠染色体中。请注意Belheouane和Srinivas的研究是关于皮肤微生物组QTLs。
右半部分显示了人类GWAS中的基因,包括UK双胞胎、FoCus/PopGeen(均为原创性研究,之后方法有修改)、LifeLines-Deep和GEM研究。
中间的连接部分显示了可能不仅在人类GWAS中出现还位于小鼠QTLs置信区间的重叠基因,它们在显示微生物组差异间的关联方面互相支持。
Benson等在2010年发表了第一项概念验证研究,显示了在小鼠的几代杂交种中,我们确实能够在特定的染色体区域将基因差异定位到肠道微生物组差异。
甚至当解析度不够高的时候,仍有一些潜在的和特定微生物组丰度相关的基因的迹象,尤其是Toll样受体2 (Tlr2)的下游基因,这个基因主要负责感知革兰氏阳性菌和下游基因(包括Irak3, Lyz2, Lyz2, IL-22和IFN-gamma),而相关的微生物组特征是红蝽菌科 (Coriobacteriaceae) 和乳球菌属 (Lactococcus)(Benson等,2010)。
McKnite等(2012)和Leamy等(2014)跟着使用不同的杂交方案,在不同的研究中鉴定出更多的带有有限重叠的区域,每个都带有与可能相关的基因有关的有趣发现。
Wang等(2015)发表了另一项有趣的研究,他们使用杂交小鼠作为QTL杂交种群,在这个种群中,家鼠的两个亚种被杂交到第二代,并且许多区域都被发现和微生物多样性相关。
实验室小鼠本质上都是Mus musculus domesticus,而其欧洲东部的同类则是Mus musculus musculus。它们在欧洲中部野生环境中自然出现,且具有被深入研究的进化和物种形成体系。现在,微生物组似乎也受这种杂交的影响。
另外,关联的类型也很有趣。有一半的关联是超亲遗传的,意味着针对一个特定基因位点的杂合子有异常高或低的值,超出纯合子的范围 (Wang等,2015)。
这和一个遵循杜布赞斯基-穆勒不相容模型的潜在上位相互作用,一起更深入的证明微生物组可以改变宿主基因组的进化。细节可以在Bodenstein研究组发表的文献和相关综述中找到 (Brucker和Bordenstein, 2013; Bordenstein和Theis,2015)。
两个皮肤微生物组QTLs均由Baines研究组组发表,他们观察了身体表面宿主-微生物组的动态平衡 (Srinivas等,2013;Belheouane等,2017)。
通过研究一种叫大疱性表皮松解症 (EBA) 的自身免疫皮肤病模型,该研究组将先前针对疾病的QTLs延伸到包括微生物组部分,发现微生物组的确在确定疾病症状中起重要作用。
甚至即使有大致相同的基因组成,是否发展成疾病和葡萄球菌属 (Staphylococcus) 的丰度高低相关,这可能是一个保护性菌种。相反,当把细菌丰度算进统计模型中时,只要控制好“噪音”或细菌的环境混淆因素,QTL的效力就会显著提高 (Srinivas等,2013)。
第二个皮肤QTL研究开创了新的研究方法,并使用16S rRNA基因转录子,检验了微生物组的“活跃”部分而非常驻菌落。再结合近一步的跨世代杂交(第15代而非第4代),结果几乎达到了单基因解析度,而且当转录子被使用时,得到更显著的关联性。
另外,一些基因位点涉及皮肤的癌变,它们和也能导致结肠癌症的类似细菌相关(Belheouane等,2017)。
我们需要注意,以上讨论的Org等(2015)的研究实际上是利用了类似在人体中应用GWAS的方法,在这个过程中,有几个和微生物分类相关的重要基因被鉴定出来。
和以上的QTLs相反,他们使用了常用的小鼠品系(共110种),而非特意设置的杂交品系,并且研究方法仔细考虑了种群结构。
这里唯一的局限性就是相对较小的样品数量和较少的SNPs,因此我们无法达到类似于在人体中的基因组显著性阈值。这限制了结果的解析度。
巧合的是,迄今为止,关于在人体中的微生物组的GWAS分析,同样也有6个例子。我们认为,其中至少有2个研究的样本量不够大,因此不能和剩余的几例同等考虑。
第一个近似于微生物组GWAS的研究,并非经由研究设计得来。Blekhman从HMP的宏基因组原始数据中提取人体基因组读数,从这些读数中提取SNPs对应于每个受试者,然后寻找宿主的基因差异和微生物组差异之间的对应关系。
一个特定关联存在于乳糖酶 (LCT) 基因和双歧杆菌属 (Bifidobacterium)之间,它们均和牛奶的摄入相关,因此可以理解是相关的 (Blekhman等,2015)。但是,“钓出”的人体读数是否足以提取SNPs并不确定,其后续分析的可靠性也不清楚。
Davenport等(2015)报道了一个规模较小但更基于传统设计的GWAS研究,并成功找到了一些关联,但它们中没有一个达到通常认可的全基因组范围内有意义的临界点(也就是5E-08或1E-08,原理是当你检查上百万个SNPs时,真正的有意义的关联应该是在经过为了多重检测而进行的Bonferroni或Benjamini-Hochberg校正后仍有意义,因此通常被设置在这个基础上)。
Ley研究组也继续他们在英国双胞胎群组和多个模型中寻找微生物组-SNP关联的研究,他们确实发现了一些有意义的地方,并且后来又再次被发现有意义,包括LCT和SLIT3。
但是,因为缺乏对研究模型或功能性研究的聚焦,研究并没有深入到足够的细节,在基因组尺度去探索基因-微生物组的纽带 (Goodrich等,2016) (图3)。
在人体GWAS领域,关于肠道微生物组的真正突破出现在2016年的Nature Genetics杂志上,来自德国(PopGen/FoCus) (Wang等,2016)、荷兰(LifeLines-Deep) (Bonder等,2016) 和加拿大的研究团队 (GEM) (Turpin等,2016) 同时发表了关于人体肠道微生物组的大规模GWAS研究(图3)。
3个研究团队都纳入了超过1000个不相关个体,全部具有验证队列,而且都考虑了细菌种类的分布特征(只有一小部分符合正常的分布,其余的主要是零膨胀zero-inflated)。
在这三项研究中,德国和加拿大团队的研究使用了一种二元栅栏模型(twopart hurdle model)来解决零膨胀,而荷兰团队研究的是非零部分(核心OTUs)。
另一个不同之处是,德国/加拿大团队研究的是基于16S rRNA的细菌组成,而荷兰研究还包含了鸟枪法宏基因组数据,因此能够标示特定的功能通路。
除了使用细菌丰度作为主要的研究特征,德国的研究还特别提出了一种方法,可以将微生物的总体多样性(β-多样性)和人体基因组的变化关联起来,他们发现了42个超过有意义临界点的基因位点,包括一个已知的涉及胆汁酸感知和稳态的维生素D受体 (VDR)。
另外,在这项研究中,包括胆汁酸分析、宏基因组测序、与不同数据库交叉检测以及比较人类转录子组和细菌丰度(“偶联”)在内的大量功能性研究,建立了VDR作为主要通过胆汁酸代谢和下游通路作用的人类-微生物组互作的有效性 (Wang等,2016)。
β-多样性关联的方法被进一步发展成需要更少的计算强度并能广泛应用于高维度的方法,在此过程中还有一些有趣的基因位点的发现 (Ruhlemann等,2017)。
荷兰的研究主要确认了先前LCT-双歧杆菌属关联度的发现,并证明了环境影响(在这里是指牛奶摄入)也和个人的基因型相互作用,并影响微生物丰度 (Bonder等,2016)。
Benson总结了这三份研究,也发表在同一期Nature Genetics上(Benson, 2016)。另外,Kurilshikov和Zhemokova也写了关于这个主题的出色综述(Kurilshikov等,2017)。
结论
我们已经介绍了在理解宿主-微生物相互作用方面基因组学研究历程,以及不同研究方法的主要结果。我们已经在比较性研究中看到了非直接证据,但那些研究具有局限性。我们能够探究感兴趣的单个基因,并深入了解它们的重要性,但并不能得到最完整的信息。
另外,现在已经有了数量遗传学的方法,其间也有需要我们小心对待的方方面面。然而,尝试远没有结束。我们还处在研究的早期阶段。这里,我们也要注意我们所提及研究的局限性,以及我们对未来努力方向的想法。
局限性
我们的综述主要集中在人类和小鼠的研究中,而忽略了其它的模型或非模式生物。原因是我们已经在前两者中花费了巨大的精力,并且人类和小鼠与我们自身的健康息息相关。
我们确实了解很多关于植物基因-微生物相互作用的文献,很多是教科书级模型,比如那些涉及农杆菌 (Agrobacterium) 侵染和定植的基因,它们所涉及的复杂相互作用,可能会让一些在动物中的宿主-微生物互作相形见绌 (Nester, 2015; Ellis, 2017)。
类似地,已有利用拟南芥发表的文章介绍关于植物领域的微生物组GWAS研究,其中也列出了可能会参与到更广泛互动的一系列基因 (Horton等,2014)。然而,因为动物中并没有许多类似的基因,或至少不存在相同的功能,它们作为其他研究模型参考物的价值有限。
我们不缺乏在秀丽隐杆线虫 (C. elegans)、果蝇 (Drosophila) 和斑马鱼等常见的实验室模式动物中宿主-微生物互作的例子。它们中的大部分都是单个致病菌,并且其结果中的差异主要归结于宿主基因的差异。
这也属于基于候选基因的方法,因为单个基因是研究的主要焦点,对那些生物中宿主-微生物的互作也有了更宽泛的了解。
讲到在微生物组中进行全基因组范围的数量遗传学研究,现已有两项研究在果蝇中进行(Chaston等,2015;Dobson等,2015),作者指出了营养成分和宿主之间的相互作用,以及菌群作为中介对营养作用实际发生的重要性。
转换到广泛应用于人类或小鼠研究中常用的说法就是,微生物组主要决定宿主的代谢组成分和之后的健康状态。
另外,还有许多其它研究,甚至包括一个针对鸡的研究 (Zhao等,2013),如果我们遗漏了其它使用不同生物的类似研究,敬请谅解。
所有的这些研究都对这一领域做出了贡献,并且我们通过拼凑这些研究得到了更多的信息,也离解开谜团更近了一步。要达到这个目标,数据的积累和方法的创新都是必须的。
当然,相关性不代表因果关系,这是关联性研究中的限制。
功能性验证和真实因果关系的确认仍处于众多基因-微生物相互作用的瓶颈期。并且,相比我们在宿主方面得到的有限知识,我们对于和宿主互作的细菌基因一无所知。
在致病菌中,我们研究了关键的病毒因子,它们是侵染过程或致病化过程的一部分,这些病毒因子包括不同的毒素、不同种类的分泌系统或产生影响宿主的关键代谢物基因。
我们也知道几个可以被宿主识别的重要分子,比如细胞壁成分——脂多糖(LPS)。但是,我们缺乏细菌基因组的哪个部分对应于建立并维持和宿主的联系的信息,以及哪个部分影响该稳态失调的信息。
身为作者,我们认为参与其中的每种细菌的角色当然都不一样,对于这些功能,环境中的细菌会需要更少的基因,而共生细菌应该会需要提供其基因组中的关键部分来行使功能;否则,它们不会和宿主维持共生的关系。
肠道微生物组、皮肤微生物组和其它部位的细菌是中介,在某种意义上来说,它们并非出于严格意义上的共生状态,但是仍然需要贡献一部分基因组来参与。
一些研究显示了昆虫中长期的细胞内共生体,已经在进化过程中丢失了它们的大部分基因组,仅仅保留了一小部分的关键基因 (Wernegreen, 2002; Bennett等,2014)。这个是否发生在肠道或皮肤是不得而知的,作者认为,如果存在,这种基因组减少的说法将被用来解释为何通过母系传播的基因会多于通常从环境中获得的基因。
展望
正如我们在本综述开篇时提到的,病原体是宿主种群中等位基因频率变化的驱动力,而且这个选择的结果是我们通常可以观察到的。但是,这极少实际发生,并且我们还没有实现可深入检测身体状况及代价的具体参数。
但是,Vallier等(2017)实施了一个惊人的研究,结果显示,在西部家鼠 (Mus musculus domesticus)的自然种群中 ,B4galnt2基因的两个等位基因同时作为长期均衡选择的结果同时存在,其中一个等位基因能够保护宿主免受不同病原体侵害,因此受到病原体驱动选择的青睐。
但是,这也导致了胃肠道的出血,并可能降低宿主的健康水平(这和被称为I型类血友病的人类出血性紊乱类似,并且也能够因为在病原体感染过程中的有益作用而保留)。从地理分布模式结合人类历史来看,这个均衡选择是相当近期的活动,作者建立了进化模型,估计了出血性等位基因的健康代价。
它显示现在观察到的等位基因频率和分布,只在出现杂合子优势的时候可以被解释,带有出血等位基因的纯合子和出血的健康代价,则有一半算作是病原体感染引起的。
当然,这不是生物学相关的证据,因为健康代价和感染代价都是极难定量的。但是,它显示了选择微生物的重要性、微生物对我们基因组微弱的改变,甚至导致如果不出现就会危害人类种群的等位基因。
这不是单一的案例,因为许多自体免疫紊乱和代谢综合症的潜在基因/等位基因被认为是病原体在过去选择的结果,它们将在未来与我们的基因组作用,并改变我们的基因组 (Nielsen等,2007;Novembre和Han, 2012; Milot和Pelletier,2013)。
到此,我们的综述主要集中在个体基因上,而且我们仅能够将它限制到主要的概念验证研究中。
类似其他任何的基因学研究,一个在理解宿主-微生物互作时的重要概念是基因-环境相互作用(G by E),当环境情况变化时,基因的情况表明了其不同作用。这已经在日常乳制品的摄入作为环境背景(Bonder等,2016)的LCT基因和双歧杆菌属的案例中显示出来(Blekhman等,2015;Goodrich等,2016)。
但是,我们没有大量其它案例,因为环境影响的内容是非常宽泛的,并且许多研究的数量不够。同时,样品数也通常限制了这种分析。另外,因为面对大部分的复杂性状,利用单个基因来解释微生物组多样性和功能的力度有限,得出的结论也只会是片面的、有误导作用的,所以现在急需多个单个基因的研究。
将多基因变化及其权重结合到一起,可以导出并用于许多疾病的多基因评估中(Dudbridge, 2013),将其用于微生物组研究可以解释单个类群或整体多样性的潜在遗传结构,并得出一个对宿主-微生物互作更完整的概述。
另一个重要的方向是由单个基因转向参与并检验微生物和特定细胞处理/信号产生的生物通路。这需要许多单个基因的富集分析(Ramanan等,2012)。
总而言之,这个引人入胜的研究领域,已经在理解基础生物学还有医学和人体健康相关应用方面展现出潜力,同时还有很多方面有待研究。
(翻译全文结束,参考文献请见pdf原文,下载地址:https://link.springer.com/article/10.1007%2Fs13238-018-0532-9 )

猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”

