IDBA-UD:组装非均匀覆盖度的宏基因组和单细胞数据
文章目录
- IDBA-UD:用于深度非常不均匀的单细胞和宏基因组测序数据的组装软件
- 热心肠日报
- 摘要
- 动机 Motivation
- 结果 Results
- 可用性 Availability
- 方法
- 图1. IDBA-UD的流程图
- 图2. 在局部装配中重建缺失k-mer的示例
- 图3. 通过迭代解决从k到k + 1的重复的示例
- 结果
- 表1. 模拟 *L.delbrueckii*(〜1.85 Mb)的100×长度为100且1%错误率读长的错误校正结果
- 表2. 组装结果以模拟的深度为10×、长度为100的*L. plantarum*(〜3.3 Mb)1%的错误率的读长
- 表3. 表2中同一数据集上IDBA-UD、IDBA的已解析分支的预期数目以及所有组装程序的实际数目
- 表4. 基于大肠杆菌单细胞的数据
- 表6. 在植物乳杆菌(〜3.3 Mb),德氏乳杆菌(〜1.85 Mb)和罗伊氏乳杆菌F275 Kitasato(〜2 Mb)的模拟宏基因组数据集上的组装结果
- 表7. 组装来自NCBI的人体肠道微生物短读长数据(SRR041654和SRR041655)的结果
- 表8. 不同分类级别的模拟宏基因组数据集上的组装结果
- 讨论与结论
- 猜你喜欢
- 写在后面
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-4QAfK82M-1570630715653)(http://210.75.224.110/Note/LiuYongXin/120411Bioinfo/0.jpg)]
IDBA-UD:用于深度非常不均匀的单细胞和宏基因组测序数据的组装软件
IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth
Bioinformatics, [4.531]
2012-4-11 ORIGINAL PAPER
DOI: https://doi.org/10.1093/bioinformatics/bts174
第一作者:Yu Peng
通讯作者:Henry C. M. Leung [email protected]
其它作者:S. M. Yiu和Francis Y. L. Chin
作者主要单位:
中国香港薄扶林道,香港大学计算机科学系(Department of Computer Science, The University of Hong Kong, Pokfulam Road, Hong Kong)
热心肠日报
IDBA-UD:宏基因组和单细胞组装工具
- IDBA-UD算法专门用于组装来自具有不均匀测序深度的单细胞或宏基因组测序技术数据;
- 使用多个深度相关阈值来去除低深度和高深度区域中的错误k-mer;
- 使用具有末端信息的局部组装技术来解决低深度短重复区域的分支问题;
- 在模拟和真实数据集上的实验结果表明,IDBA-UD在深度高度不均匀的数据集中的表现优于所有现有的组装程序(SOAPdenovo、Velvet和Meta-IDBA等)。
点评:IDBA-UD是目前宏基因组最常用的三款拼接软件之一,另外两个分别为拼接结果最快的MEGAHIT(http://www.mr-gut.cn/papers/read/1051360183 )和最长的metaSpades。IDBA—UD的速度和内存消耗介于两者之者,是平衡的选择,优点也较多,可实现从小到大迭代k,还通过局部组装重建缺失的k-mers,并通过迭代地去除低深度重叠群来去除错误。最近Nature Biotechnology的瘤胃文章(http://www.mr-gut.cn/papers/read/1066063689 )也选用了些方法组装。
摘要
动机 Motivation
下一代测序使我们能够使用单细胞测序或宏基因组测序技术对微生物环境中进行测序。 但是,这两种技术都存在以下问题:基因组的不同区域的测序深度或来自不同物种的基因组的测序深度高度不均匀。大多数现有的基因组组装软件通常会假设测序深度是均匀的。 这些组装程序无法构建正确的长重叠群。
结果 Results
我们介绍了基于de Bruijn图方法的IDBA-UD算法,用于组装来自具有不均匀测序深度的单细胞测序或宏基因组测序技术获得的读长。 已经采用了几种创新的技术来解决这些问题。 代替使用简单的阈值,我们使用多个深度相关阈值来去除低深度和高深度区域中的错误k-mer。 使用具有末端信息的局部组装技术来解决低深度短重复区域的分支问题。 为了加快处理过程,执行了一个纠错步骤,以纠正可与高置信度重叠群对齐的高深度区域的读长。 比较IDBA-UD和现有组装软件(Velvet,Velvet-SC,SOAPdenovo和Meta-IDBA)对不同数据集的性能,结果表明IDBA-UD可以以更高的精度重构更长的重叠群。
可用性 Availability
http://www.cs.hku.hk/~alse/idba_ud
方法
图1. IDBA-UD的流程图
Flowchart of IDBA-UD
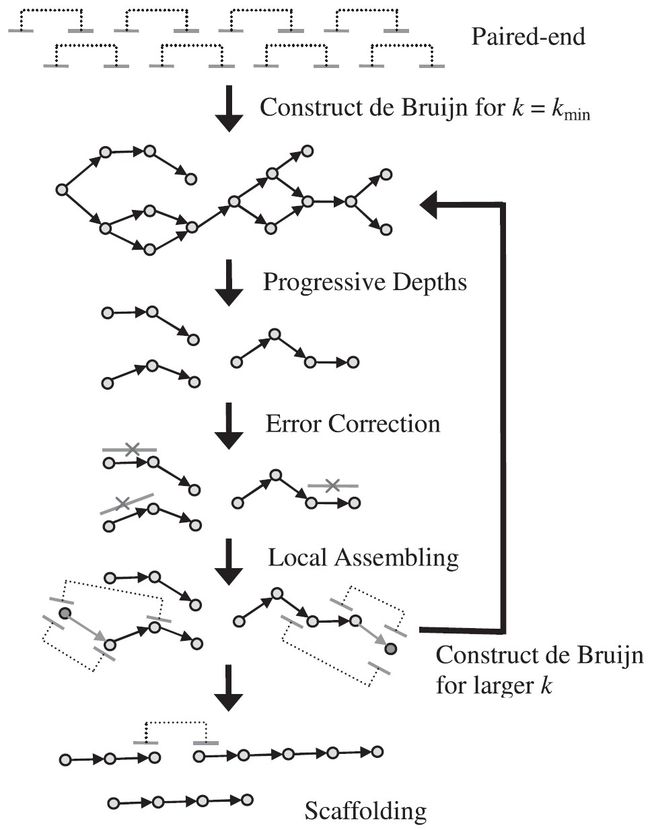
IDBA-UD的主要步骤流程图如图1所示。IDBA-UD将k的值从kmin迭代为kmax。在每个迭代中,固定的k的“累积de Bruijn图” Hk由输入读长和先前迭代中构建的重叠群(Ck-s和LCk-s)的集合构成,即这些重叠群被视为针对HK在每次迭代中的组装,IDBA-UD也会逐渐增加低深度切断的阈值,以去除一些低深度重叠群,从而获得更长的Hk置信重叠群(Ck)。通过使读长与一些可信的重叠群对齐,可以纠正读长中的错误。可以通过局部组装成对的末端读长(末端与一个高质量的重叠群对齐),重建的那些重叠群(LCk)中恢复一些缺失的k-mer。这些缺失k-mer的信息将通过这些重叠群(LCk)传递给下一个迭代,以构建Hk + s。最后,所有输出的重叠群均用于通过配对末端读长信息形成支架(scaffolds)。
图2. 在局部装配中重建缺失k-mer的示例
Example of reconstructing missing k-mer in local assembly.
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-7ZYzUHW4-1570630715654)(http://210.75.224.110/Note/LiuYongXin/120411Bioinfo/2.jpg)]
ACT是基因组中的一个重复区域,没有包含AACTG或TACTT的读段用于解析重复分支。 在本地组装中,ACT不再是重复,因此可以从本地读长中重建覆盖AACTG的简单路径(本地重叠群)
图3. 通过迭代解决从k到k + 1的重复的示例
Example of resolving repeats by iteration from k to k+1

重复区域是单个k-mer,uvw和u’vw’出现在基因组中,迭代后,解决重复的v
结果
表1. 模拟 L.delbrueckii(〜1.85 Mb)的100×长度为100且1%错误率读长的错误校正结果

表2. 组装结果以模拟的深度为10×、长度为100的L. plantarum(〜3.3 Mb)1%的错误率的读长
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-KqNXO4m1-1570630715656)(http://210.75.224.110/Note/LiuYongXin/120411Bioinfo/t2.jpg)]
IDBA-UD有最好的N50和覆盖度
表3. 表2中同一数据集上IDBA-UD、IDBA的已解析分支的预期数目以及所有组装程序的实际数目

IDBA-UD最大限度的解决分枝问题
表4. 基于大肠杆菌单细胞的数据
The assembly results on real single-cell sequencing data of E.coli (~4.64 Mb)

读长长度为100 nt,插入距离为〜215 nt,平均深度为〜600×。IDBA-UD有最长的N50和覆盖度。
表6. 在植物乳杆菌(〜3.3 Mb),德氏乳杆菌(〜1.85 Mb)和罗伊氏乳杆菌F275 Kitasato(〜2 Mb)的模拟宏基因组数据集上的组装结果
The assembly result on simulated metagenomic dataset of L. plantarum (~3.3 Mb), L.delbrueckii (~1.85 Mb) and L.reuteri F275 Kitasato (~2 Mb)
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-MWwDKhq0-1570630715657)(http://210.75.224.110/Note/LiuYongXin/120411Bioinfo/t6.jpg)]
这些物种的测序深度分别为10倍,100倍和1000倍。 读取长度为100,错误率设置为1%,插入片段大小遵循正态分布N(500,50)。
表7. 组装来自NCBI的人体肠道微生物短读长数据(SRR041654和SRR041655)的结果
The assembly results on human gut microbial short read data (SRR041654 and SRR041655) from NCBI

读取的长度为100,插入片段大小为260。
表8. 不同分类级别的模拟宏基因组数据集上的组装结果
The assembly results on simulated metagenomic datasets in different taxonomic levels
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-PABlI2Qz-1570630715658)(http://210.75.224.110/Note/LiuYongXin/120411Bioinfo/t6.jpg)]
对于每个级别,将生成具有三个随机选择的物种进行测试。 三种物种的深度分别设置为10倍,50倍和250倍。 读长为100,错误率设置为1%,插入距离遵循正态分布N(500,50)。 该表中显示的值是平均结果合格的测试使用示例。
结果表明IDBA-UD有着最长的N50,比Meta-IDBA高10倍以上,比SOAP和Velvet高上百倍。而且错误率也极低。
讨论与结论
在本文中,我们提出了一种新的组装程序IDBA-UD,它是IDBA的扩展,用于组装深度高度不均匀的短测序读长。 除了从小到大迭代k,IDBA-UD还通过局部组装重建缺失的k-mers,并通过迭代地去除低深度重叠群来去除错误。 在模拟和真实数据集上的实验结果表明,IDBA-UD在深度高度不均匀的数据集中的表现优于所有现有的组装程序。 对于宏基因组数据,来自同一物种的亚种的基因组之间的通用kmer比来自不同物种的基因组更为常见。 此信息在Meta-IDBA中用于组装宏基因组学数据。 作为将来的工作,我们应该研究如何将这些信息集成到IDBA-UD中以获得更好的性能。
原文链接:https://academic.oup.com/bioinformatics/article/28/11/1420/266973/
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-HHLnMJ9V-1570630715658)(http://bailab.genetics.ac.cn/markdown/life/yongxinliu.jpg)]
学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”

[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-zZsxk2PD-1570630715659)(http://bailab.genetics.ac.cn/markdown/train/1809/201807.jpg)]
点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA