中国核酸数据库GSA数据提交指南
文章目录
- 中国核酸数据库GSA
- GSA 数据模型
- 数据触发机制说明
- 发布策略和免责声明
- 如何提交数据到GSA?
- 第一步: 注册账户
- 第二步:进入GSA数据库创建GSA
- 1. 创建项目(BioProject)
- 2. 创建样本(BioSample)
- 3. 构建GSA数据集
- 数据追踪
- 引用格式
- 附录
- 计算MD5值
- 官方推荐不同平台使用的工具
- 压缩fq文件
- 提交数据文件如何命名?
- ftp提交数据
- 猜你喜欢
- 写在后面
中国核酸数据库GSA
https://bigd.big.ac.cn/gsub/
GSA 数据模型
为确保与国际同类数据库系统的兼容性,GSA遵循INSDC联盟的数据标准,GSA元数据类别主要包括项目信息(BioProject,归档于生物项目数据库)、样本信息(BioSample,归档于生物样本数据库)、实验信息(Experiment)、以及测序反应(Run)信息。项目信息是用来描述所开展研究的目的、涉及物种、数据类型、研究思路等信息;样本信息是指本研究涉及的生物样本描述,如样本类型、样本属性等;实验信息包括实验目的、文库构建方式、测序类型等信息;测序反应信息包括测序文件和对应的校验信息。各类数据之间采用线性、一对多的模式进行关联,从而形成“金字塔”式的信息组织与管理模式(图1)。

数据触发机制说明
数据发布时,相关的BioProject、BioSample与GSA数据集遵循以下触发机制(图5):
- BioProject发布不会触发相关联BioSample信息与GSA数据集释放;
- GSA数据集发布,会触发相关联BioProject和BioSample信息释放。
因此,请慎重填写BioProject、BioSample与GSA “发布时间”,一旦发布就代表数据或信息可供其他用户公开检索或下载。
发布策略和免责声明
-
用户可自行设定“发布日期”,在该日期之前,GSA保证数据不公开;
-
“发布日期”可以在GSA提交系统内进行修改:https://bigd.big.ac.cn/gsub/submit/gsa/[substitute your GSA accession number]/contents
-
如果引用这些数据和该accession号的文章先于您设定的发布时间而发表,我们将根据文章的发表时间来发布该数据;否则GSA将根据您设定的发布日期而发布该数据;
-
一旦文章发表,数据可以发布,请把已发表文章的全部信息–作者,题目,期刊,刊号,页数,日期信息发送到该邮箱: [email protected]
如何提交数据到GSA?
注册用户可通过中心生物数据统一汇交入口——生物数据递交系统(BIG Submission,BIG Sub,https://bigd.big.ac.cn/gsub/)进行一站式数据递交,具体步骤请查阅GSA使用说明。
下面我使用自己的数据来演示如何提交二代测序数据
第一步: 注册账户
如何开始提交数据?账户注册完成后,您可遵循以下原则进行数据信息录入:
这里没有什么注意的,只是填写的信息比较多,我们选择性的将带有号的信息填写上就可以了,注意全部为英文。
第二步:进入GSA数据库创建GSA
这里主要有三个步骤:
- 1.创建项目(BioProject);
- 2.创建样本(BioSample);
- 3.创建GSA数据集;
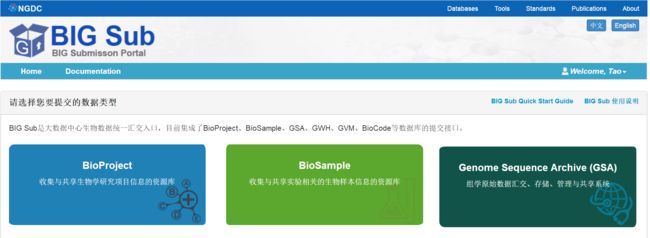
1. 创建项目(BioProject)
如果您之前没有创建项目(BioProject)请进入 BioProject 数据库完成创建:
- 按照图示步骤点击:项目(BioProject)的构建分为五个步骤,第一个是提交者信息,这里一般不需要修改,直接点击保存即可;
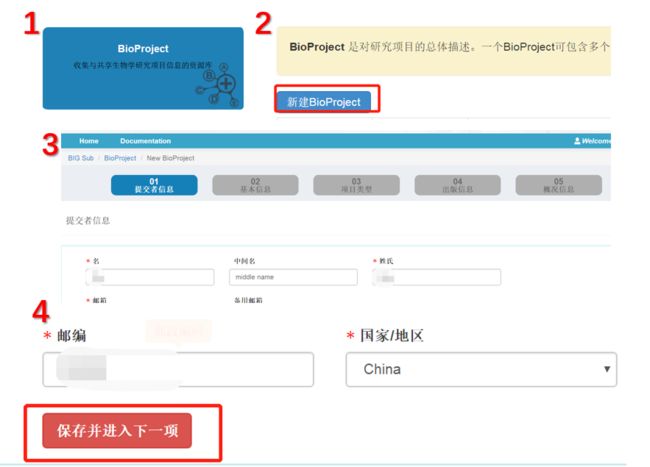
-
第二个是基本信息:我们需要指定发布的日期,这里选择日期,不同于NCBI的是在我们公开的日期之前,数据可以随时修改时间,很是方便;其次一次填写图中标记的必填模块,注意6,这里项目批准号如果没有请填写:N/A即可。
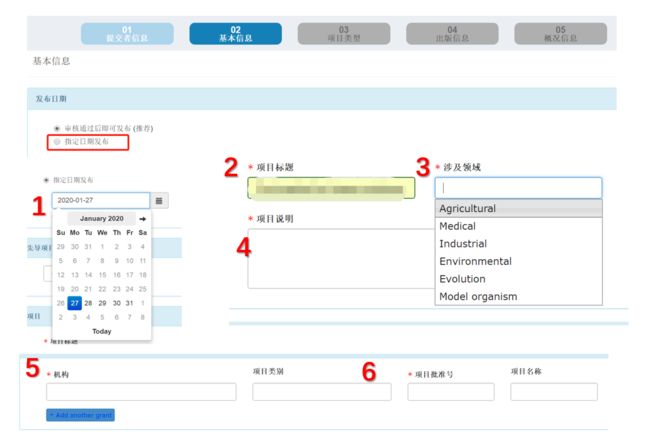
-
第三个是项目类型:这里我们需要操作两个选项,第一个是序列类型,这里我选择的是metagenome,其次是样本范围,选择:env;

- 第四个出版信息:在出版后在公布数据相信已经很少了,很多出版社都要求在投稿就要长传数据了,所以这里我们也没什么好填写的:
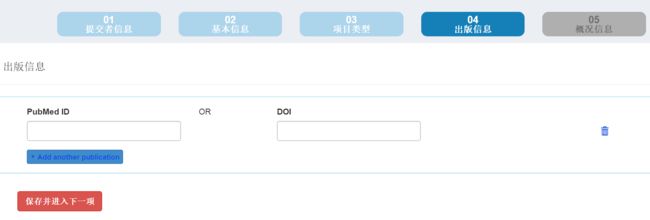
- 第五个就是概况信息;这里我们检查前面几个填写的内容即可,如果发现前面有内容不对,直接点击这五个模块中的任何一个都可以转到该模块进行更正。很方便。
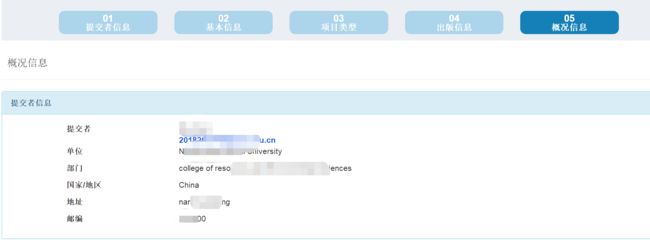
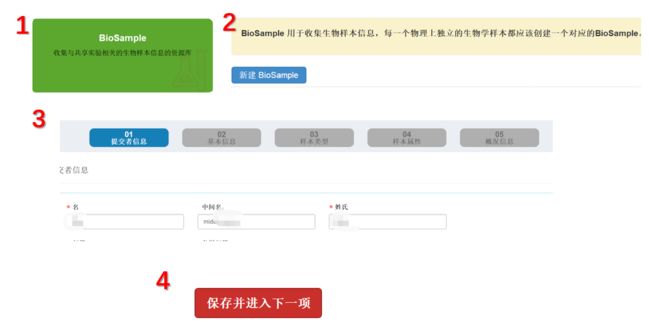
2. 创建样本(BioSample)
如果您之前没有创建样本(BioSample)请进入 BioSample 数据库完成创建:
-
第一步都一样,是确认提交者信息,一般不做修改:
-
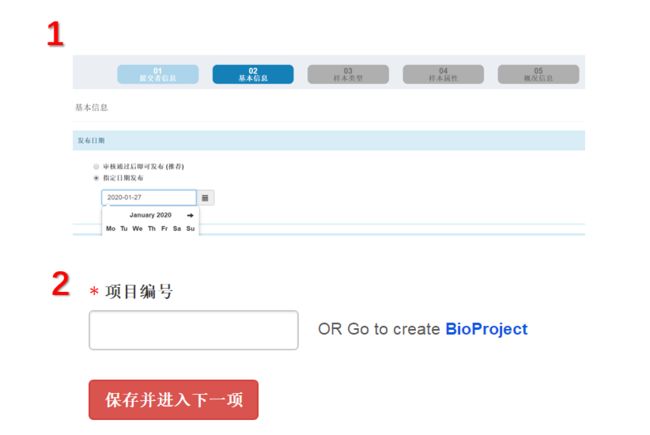
第二步 选择时间和项目同一个试时间即可,输入项目号,注意项目号就是上面我们填写完成后生成的。
-
第三步:填写样本类型,选择一个即可,都是中文注解,所以还是很容易明白的;保存进入下一步即可。

- 第四步也是最重要的一步,填写样本文件:这里有一个示例文件,我们下载:
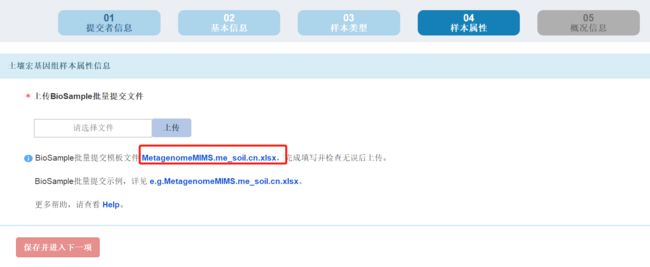
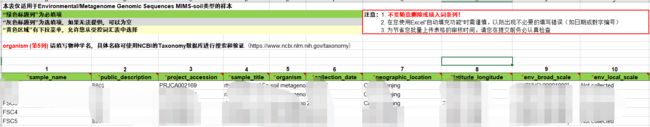
这里我认为直接看文件的解释就够了,因为都是中文的:但是为了避免大家出问题,我重点说一下这几个列:geographic_location:采样地点,这个一定要按照要求填写,例如:China: Nanjing;还有:latitude_longitude是经纬度例如32.03N 118.84E; 样本注解:env_broad_scale,这里我是土壤,所以套入格式:soil [ENVO:00002007]
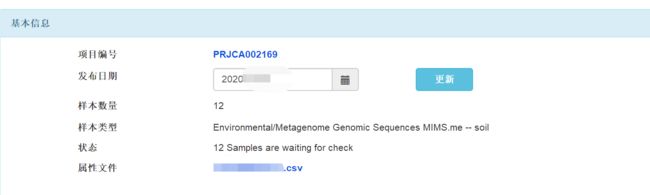
- 第五步 查看我们填写的信息即可;
3. 构建GSA数据集
完成 GSA数据集中Experiment和Run的元数据信息录入——实现与BioProject、BioSample和数据文件的相互关联。通过FTP完成数据文件上传。
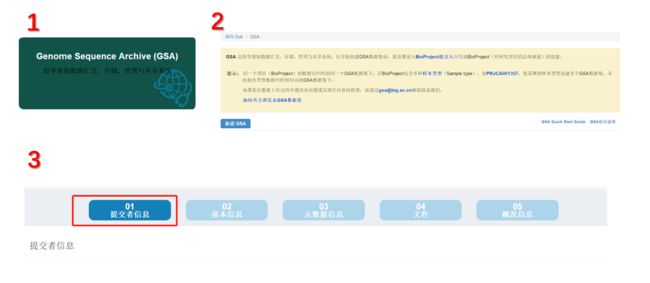
- 第一步: 点击GSA按照图中顺序进行点击,第一个提交者信息和前面两个都是一样的,所以不用动保存进入第二步:
-
第二步:基本信息需要填写标题,项目号,选择已经建立sample,即可进入下一步
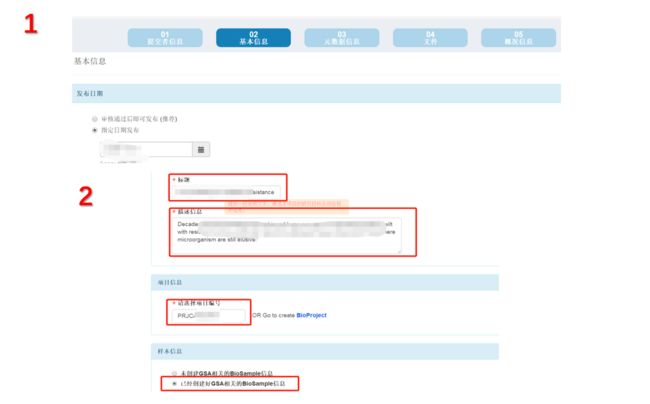
-
第三步:上传测序数据:
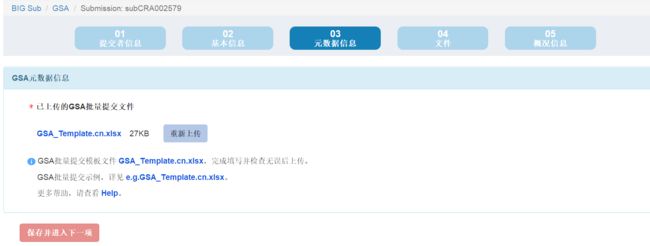
这里重点是填写序列信息,不同于NCBI的是这一步一共有两张sheet,第一张是:试验,第二章是run
首先我们填写第一张sheet:这里需要注意的就没什么了,只要你阅读完填表之前的子基本没什么问题:

其次我们填写第二章sheet:
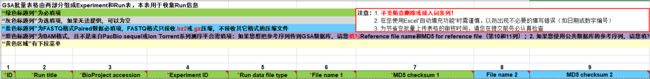
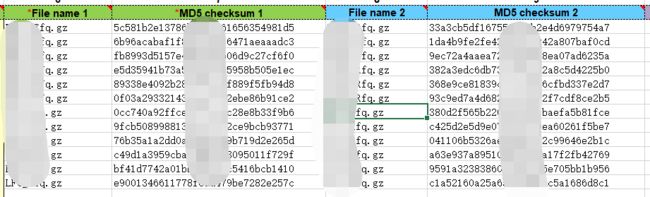
这里需要注意我们上传的必须是压缩文件;其次必须有MD5值:这里为了保证流程的顺畅,我将这些步骤的操作方法附在后面,大家可以尝试:
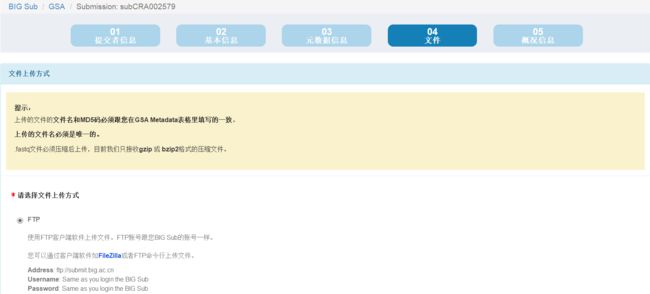
- 第四步:上传数据,这里我选择用ftp服务器上传,具体步骤见附件:
- 第五步:就可以进行查看前四步的信息了,当然可以随时修改前面的信息;

查看数据

- 第六步:上传结果修改和追踪
通常状况下,数据信息与文件审核归档约需要 1-2 天(数据量越大相应所需时间越
长),归档成功后您会收到一封通知邮件,并可在 GSA 列表中查找的为您分配的 GSA 编
号(GSA Accession number);如果归档中数据信息与文件审核归档过程中出现问题,信
息将反馈到您的注册邮箱,因此请您关注邮箱反馈信息。

注:
- GSA 提交编号(Submission ID):sub#,如上图中的 subCRA000595。请仅在
联系 GSA 工作人员时使用,不要在 BIG Search 检索信息时或在文章中使用提交
编号。 - 请务必在 BIG Search 检索信息时或在文章中使用 GSA 编号(GSA Accession
Number):CRA#,如上图中的 CRA000532。
数据追踪
由于GSC数据库管理员需要审核数据,所以一般需要等待两三天,我这批项目在过年上传的,所以等待的时间相对较长,在前天,管理员发来邮件告诉我数据MD5值不匹配。一下是邮件内容:
Dear Tao Wen,
NOTE: THIS MAIL IS SENT BY SYSTEM AUTOMATICALLY, PLEASE DO NOT REPLY IT DIRECTLY.
If you have any question, please contact [email protected]
-------------------------------
This email has been sent to inform you that your submission subCRA002580 is waiting for files:
CRR115717:
A1_F.fq.gz ef96d3342977e67954ae2643439bdbb3 The MD5 code of the uploaded file does not match the submitted value.
A1_R.fq.gz 7022033595ed7e415c8321bc30878ddb The MD5 code of the uploaded file does not match the submitted value.
自己追溯了我的流程,错误是由于计算MD5值的文件是fq,但是上传的是压缩文件,也就是说文件的压缩也会影响MD5值。
所以我删除GSA项目后重新填写上传了一份修改后的文件。
在第二天就确认成功了,发过来 邮件
Dear Tao Wen,
Your submission:subCRA002XXX is checked OK. The assigned accession of the submission is: CRA002XX, which can be cited in your publication. Thank you for submitting data to GSA.
GSA DATA ADMIN
2020-02-08 11:01:38
-------------------------------
NOTE: THIS MAIL IS SENT BY SYSTEM AUTOMATICALLY, PLEASE DO NOT REPLY IT DIRECTLY.
引用格式
这一套工作做完后我们就可以引用了,参考刘老师NBT引用格式:
Raw sequence data reported in this paper have been deposited (PRJCA001214)
in the Genome Sequence Archive in the BIG Data Center, Chinese Academy
of Sciences under accession codes CRA001372 for bacterial 16S rRNA gene
sequencing data and CRA001362 for metagenomic sequencing data that are
publicly accessible at http://bigd.big.ac.cn/gsa.
注意:引用如下文章:Members, B. I. G. D. C. Database resources of the BIG Data Center in 2018.
Nucleic Acids Res. 46, D14–D20 (2018).
附录
计算MD5值
官方推荐不同平台使用的工具
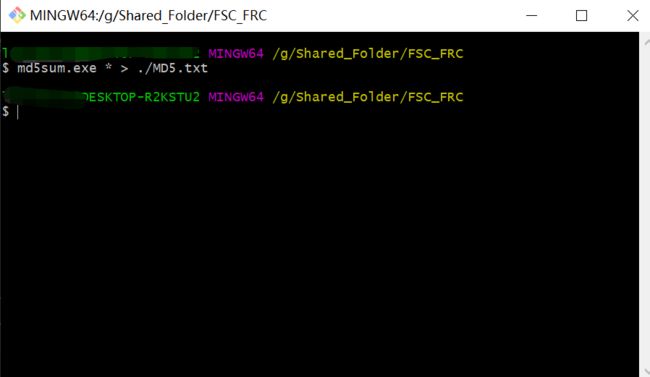
- Linux 用户请使用 $ md5sum 命令计算;
- Mac 用户请使用$ md5 命令计算;
- Windows 用户请使用第三方工具进行计算,例如 winmd5free http://www.winmd5.com/。
本来我在win上操作,所以就下载了winmd5free,但是只能一次压缩一个文件,这样我就不想用了。但是在家里没有linux平台,远程还需要传输数据,所以我就选择了xshell,这个工具已经集成进去了,我们可以直接使用,并且可以使用通配符,我们一起搞定啦。
压缩fq文件
中国核酸数据库需要提供压缩文件的fq格式文件上传,此时我继续使用bshell工具,一条命令:gzip -c B80-1.R1.fq > B80-1.R1.fq.gz,但是我有好多,需要写个循环:
for tar in *.fq;do echo ${tar}; gzip -c ${tar} > ./${tar}.gz; done
提交数据文件如何命名?
对于提交FASTQ格式的数据,每一个RUN包含文件数请不要超过两个,即单端测序数据(Fragment)RUN文件数为一个,双端测序数据(Paired-end)RUN文件数为两个(通常单个文件不要超过10GB)。Fragment数据以单个文件上传,务必写全名称(包括文件后缀名),如:DRT_10107_1.clean.fq.gz。对于Paired-end数据,请把两个数据放在同一个Run里面,务必写全名称(包括文件后缀名),并用F和R在文件名中做标记,例如,用F和R在文件名中做标记, DRT_10107_F.clean.fq.gz;DRT_10107_R.clean.fq.gz。
完成之后我看到对文件命名 是有要求的,所以我进行文件名的批量修改,这里由于shell我命不熟悉,所以我写了R脚本进行文件名的修改:
这是代码:
# 提取文件夹下的文全部件名称
fl_1 <- dir()
#这里我们提取复合要求的指定文件名称
fl_1 =dir("./", pattern = c("B|L"), full.names = TRUE, ignore.case = TRUE)
fl_2 <- as.character(fl_1)
fl_2 = gsub("-","",fl_2)
fl_2 = gsub(".R1.","_F.",fl_2)
fl_2 = gsub(".R2.","_R.",fl_2)
fl_2
file.rename(fl_1, fl_2) #函数形式为file.rename(from, to),from为原始文件名向量,to为新的文件名向量
ftp提交数据
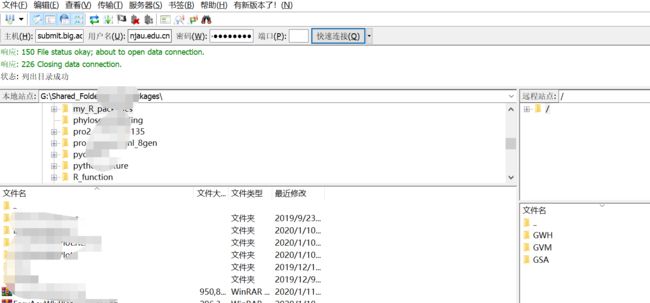
如何通过FTP连接到GSA的服务器?
请使用 FTP客户端软件(比如 FileZilla Client)登录 FTP 服务器。请采用二进制模式上传,如果是用FTP软件上传,请参考软件说明进行设置;如果是用FTP指令上传,请在“mput”指令前,先运行“binary”指令。
FTP服务器地址:submit.big.ac.cn
用户账号与BIG sub账号一致。如果不知道是什么?直接到自己的profile下去查看:
注意:用户登录自己的FTP路径后,先cd 到 /GSA目录下再上传文件。
注意地址是:submit.big.ac.cn
撰文:文涛 南京农业大学
责编:刘永鑫 中科院遗传发育所
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA