PICRUSt:16S预测宏基因组-扩增子分析锦上添花
写在前面
16S分析能获得的信息比较有限,一般找到差异OTU,就很难再深入分析了。
如何把差异OTU与细菌自身的基因组功能建立联系呢?很多人在这方面做出了努力。
PICRUSt就是让16S扩增子分析锦上添花的工具,可以基于OTU表预测宏基因组的功能基因组成。该软件2013年发表在Nature Biotechnology上,截止18年11月24日引用2094次,由Curtis Huttenhower团队开发。最新本地版本1.1.2于17年8月30日发布。在线版最新为1.1.1于4月25日发布。
已经有很多人写过PICRUSt的原理介绍,比如《根据16S预测微生物群落功能最全攻略》。
但推荐的方法再好,很多小伙伴实际操作中还是有一定困难的。“宏基因组”本着只分享干货的原则,推出扩增子预测宏基因组的中文实操指南。将介绍一系列相关软件的详细使用。
本部分练习所需文件,请在公众号后台回复“扩增子”获得。本练习是在扩增子分析流程中的可选扩展功能。原始流程阅读请点我
制作PICRUSt要求OTU表
大多数人还要用de novo方法建的OTU表,而PICRUSt需要以greengene13.5版本为参考序列的OTU,这就需要自己动手了,可以用linux服务器,也可以用qiime虚拟机搞定(一般公司测序都会给分析好,没有就找他们要)。
创建基于Greengene13.5的close reference OTU表的方法
# 进入测序数据的文件夹
cd ~/example_PE250
# 下载13.5版参考序列,QIIME中13.8版本此软件不支持
wget ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_5_otus.tar.gz
tar xvzf gg_13_5_otus.tar.gz
# Usearch产生OTU表
./usearch10 -usearch_global temp/seqs_usearch.fa -db gg_13_5_otus/rep_set/97_otus.fasta -otutabout result/gg135_otu_table.txt -biomout result/gg135_table.biom -strand plus -id 0.97 -threads 20
# 转换文本为biom标准格式
biom convert -i result/gg135_otu_table.txt -o result/gg135_otu_table.biom --table-type="OTU table" --to-json在线版操作
Picurst在线服务器 http://huttenhower.sph.harvard.edu/galaxy
准备数据和上传
先本地生成close reference OTU(gg135_otu_table.txt 或 gg135_table.biom),或用在线测序数据 https://raw.github.com/picrust/picrust/master/tutorials/hmp_mock_16S.tab
先从左侧下部get data — upload file — choose local file — type为Auto-dected或picurst;再选择start,完成后点close拷贝数标准化
左侧工具栏 PICRUSt — Normalize By Copy Number
biom格式选择第一项,文本格式选择第二项,提交即可(biom格式报错;文本的也报错。可能是服务器暂时不可用,换个时间一般会好)。预测宏基因组
左侧工具栏 PICRUSt — Predict Metagenome,
选择刚生成的Normalized By Copy Number on data xx,点击execute即可。按功能类别分类汇总
左侧工具栏 PICRUSt — Categorize by function, 输入选择Predict Metagenome on data xx, 级别使用默认的3,输出默认为BIOM,建议改为txt,点击Execute。就会生成一个表下载,这就是转换为KO的表,和OTU类似,但是功能描述。
Linux本地版使用
安装picrust
PICRUST http://picrust.github.io/picrust/install.html#install
依赖关系包括: python 2.7, PyCogent 1.5.3, biom 2.1.4, numpy 1.5.1, 这些都是qiime1.9.1所依赖的,因此只要安装过qiime这都不是问题了(all installed in qiime),详见QIIME安装
# 下载并安装,管理员权限非常顺利,也方便所有人使用
git clone git://github.com/picrust/picrust.git picrust
cd picrust
sudo pip install .
# 下载数据库
download_picrust_files.py运行picrust
# 按拷备数标准化OTU表, -c指定数据库
normalize_by_copy_number.py -i result/gg135_table.biom -o normalized_otus.biom # 用usearch的biom结果会报错
normalize_by_copy_number.py -i result/gg135_otu_table.biom -o result/normalized_otus.biom -c picrust/picrust/data/16S_13_5_precalculated.tab.gz
# 预测宏基因组
predict_metagenomes.py -i result/normalized_otus.biom -o result/metagenome_predictions.biom -c picrust/picrust/data/ko_13_5_precalculated.tab.gz
# 转换结果为txt查看
biom convert -i result/metagenome_predictions.biom -o result/metagenome_predictions.txt --table-type="OTU table" --to-tsv
# 调整为标准表格
sed -i '/# Const/d;s/#OTU //g' result/metagenome_predictions.txt
# 预览KO表,和OTU表类似,只是OTU变为了KO
less result/metagenome_predictions.txt
# 统计KO数据6910个
wc -l result/metagenome_predictions.txt
# 按功能级别分类汇总
# -c指输出类型,有KEGG_Pathways, COG_Category,RFAM三种,-l是级别,分4级
categorize_by_function.py -i result/metagenome_predictions.biom -c KEGG_Pathways -l 3 -o result/metagenome_predictions.L3.biom
# 转换结果为txt查看
biom convert -i result/metagenome_predictions.L3.biom -o result/metagenome_predictions.L3.txt --table-type="OTU table" --to-tsv
# 调整为标准表格
sed -i '/# Const/d;s/#OTU //g' result/metagenome_predictions.L3.txt
# 预览KO表,和OTU表类似,只是OTU变为了KO
less -S result/metagenome_predictions.L3.txt
# 统计KO数据L3级别有329个
wc -l result/metagenome_predictions.L3.txt
# 查找指定通路的丰度情况(可选,用于具体查看某个KO)
metagenome_contributions.py -i result/normalized_otus.biom -l K00001,K00002,K00004 -o result/ko_metagenome_contributions.tab最终结果数据格式
展示KO表的部分结果格式
ID KO4 KO6 KO5 KO3 WT8 KO9
K01365 0.0 0.0 0.0 0.0 0.0 0.0
K01364 0.0 0.0 0.0 0.0 0.0 0.0
K01361 31.0 29.0 31.0 24.0 2.0 26.0
K01360 2.0 0.0 1.0 0.0 1.0 0.0
K01362 77717.0 50358.0 66631.0 81057.0 33085.0 90576.0
K02249 5.0 11.0 4.0 4.0 0.0 2.0
K05841 3123.0 2044.0 2226.0 6404.0 1534.0 2795.0展示KO level3级的部分结果格式
ID KO4 KO6 KO5 KO3 WT8 KO9
1,1,1-Trichloro-2,2-bis(4-chlorophenyl)ethane (DDT) degr
ABC transporters 3016942.0 2108175.0
Adherens junction 142.0 154.0 123.0 119.0
Adipocytokine signaling pathway 90482.0 65434.0 76865.0
African trypanosomiasis 15284.0 10342.0 13778.0 13364.0
Alanine, aspartate and glutamate metabolism 680022.0
Aldosterone-regulated sodium reabsorption 85.0
Alzheimer's disease 85944.0 58346.0 76213.0 73664.0
Amino acid metabolism 218212.0 148475.0
Amino acid related enzymes 929584.0 651269.0
Amino sugar and nucleotide sugar metabolism 659863.0
Aminoacyl-tRNA biosynthesis 664516.0 474744.0
Aminobenzoate degradation 485766.0 339700.0宏基因组功能分类(KO)表统计与可视化
我们现在获得了KO表或L3级别的功能表,它们同OTU表类似。接下来,可以用官方推荐的STAMP、HUManN、LEfSe和GraPhlAn进行分析。
也可以采用之前我介绍的edgeR或DEseq等R包,对KO表按分组进行统计,找到差异用热图或柱状图展示即可。详见 热图:差异菌、OTU及功能
我们可以使用上文中的代码,对KO像OTU一样分析,看样品间的相关性聚类、差异KO功能分析并热图可视化。
KO与OTU相关分析比较
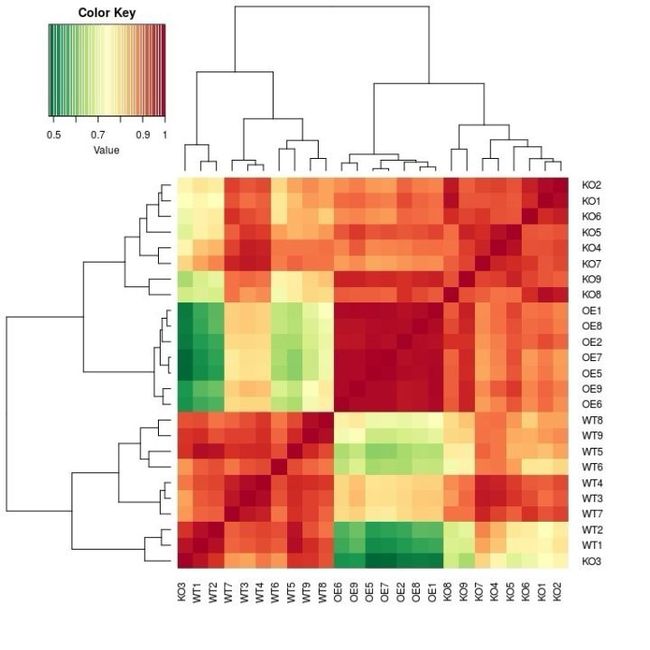
图1. 原始基于OTU相对丰度样品相关性的热图,WT, OE,KO(这里KO是组名,表示基因敲除)明显分为三组
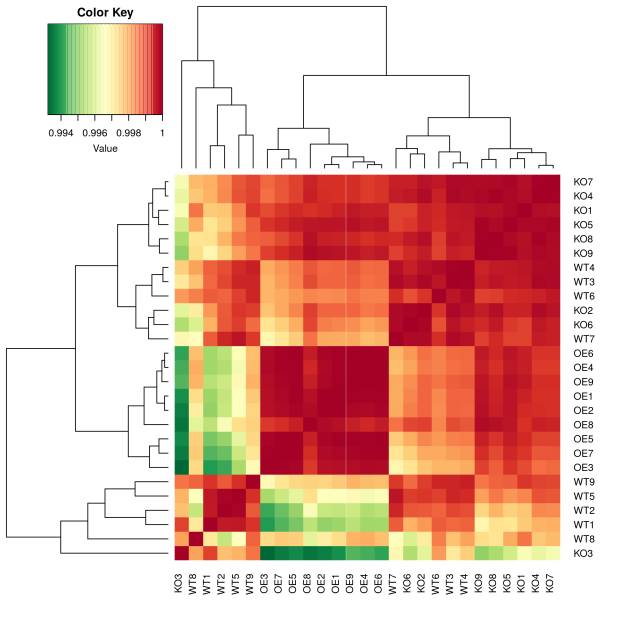
图2. 基于宏基因组功能(KO)相对丰度样品相关性热图,OE还是明显的一组,KO和OE的差别非常不明显,而且分成了两个混杂的类群。
一种可能是这个基因的突变体引起了菌群结构变化,但在功能基因层面真的和WT差异不大。但更可能的原因是:PICRUSt在本研究中预测的宏基因组数据不够准确全面,偏差到了无法区分WT与突变体,因为原始注释中有很多OTU并不在greengene数据中可以匹配,这些PICRUSt是无法分析的。
差异功能KO统计与热图展示
但OE与WT差异还是明显的,我用上文热图中的代码套用在KO表进一步统计差异KO,并展示。
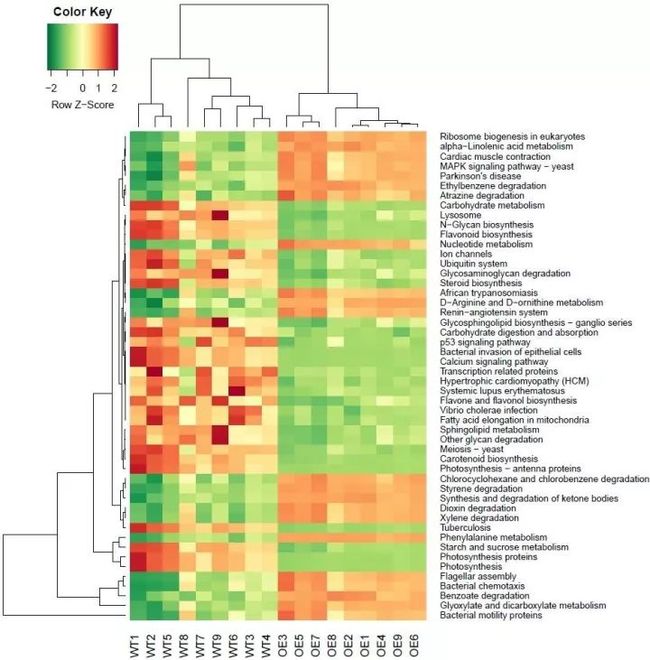
图3. 差异KO(Level3, P < 5e-7)
详细的绘图代码参考上文热图,也可以在百度云result目录中下载DAKO_egr.r文件,读取OTU替换为KO的表,绘图主要变化只有两处如下:
# 筛选差异KO的阈值,用默认的0.05会有168个差异,热图展示会看不清字。调整至5e-7减少至50个左右只是为方便展示
de_lrt = decideTestsDGE(lrt, adjust.method="fdr", p.value=0.0000005)
# 热图绘制,注意KO的名称较长,调整margins第二个值为17才显示完整
heatmap.2(sub_norm, scale="row", Colv=TRUE, Rowv=TRUE,dendrogram="both", col=rev(colorRampPalette(brewer.pal(11, "RdYlGn"))(256)), cexCol=1,keysize=1,density.info="none",main=NULL,trace="none",margins = c(3, 17))关于差异KO的展示,还常用泡泡图、柱状图结合不同分类级分组展示,这都是绘图的范畴了,我会在将来也会进行详细讲解的。想要自己画的,可以先学习陈同博士的R绘图教程(见公众号菜单-文章目录-R语言绘图部分)。
接触过泛基因组人都知道,即使16S相同,由于细菌序列的大量水平转移,功能基因也是相差极大。所以本软件的结果,仅供描述。富人做宏基因组,穷人16S预测功能锦上添花。
Reference
Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Langille, M. G.I.; Zaneveld, J.; Caporaso, J. G.; McDonald, D.; Knights, D.; a Reyes, J.; Clemente, J. C.; Burkepile, D. E.; Vega Thurber, R. L.; Knight, R.; Beiko, R. G.; and Huttenhower, C. Nature Biotechnology, 1-10. 8 2013.
原文 http://www.nature.com/nbt/journal/v31/n9/full/nbt.2676.html
主页 https://picrust.github.io/picrust/
1.1.1服务器 http://galaxy.morganlangille.com/
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2400+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读