水稻微生物组时间序列分析3-冲击图展示时间序序列变化
-
- 写在前面
- 图3. 哪些菌门随时间呈现规律变化呢?
- 绘图实战
- 清空工作环境和加载包
- 读入实验设计、OTU表和物种注释
- 筛选高丰度门用于展示
- 数据交叉筛选
- 按样品绘图
- 按组绘图
- 绘制冲击图alluvium
- 猜你喜欢
- 写在后面
写在前面
之前分享了3月底发表的的
《水稻微生物组时间序列分析》的文章,大家对其中图绘制过程比较感兴趣。一上午收到了超30条留言,累计收到41个小伙伴的留言求精讲。
我们将花时间把此文的原始代码整理并精讲,祝有需要的小伙伴能有收获。
本系列按原文4幅组图,共分为4节。本文是第二节b,散点图拟合。
时间序列分析中,流图可以非常好展示研究对象随时间的变化,如连线堆叠图、冲击图、桑击图。之前我们对这几类图都有专题介绍,如下:
- 堆叠柱状图各成分连线画法:突出组间变化
- 冲击图展示组间时间序列变化ggalluvial
- 桑基图riverplot
今天以图3中的一个子图,来实践一下冲击图的绘制。
前情提要
- 水稻微生物组时间序列分析
- 1模式图与PCoA
- 2a-相关分析
- 2b-散点图拟合
先了解一下图2的内容。
图3. 哪些菌门随时间呈现规律变化呢?
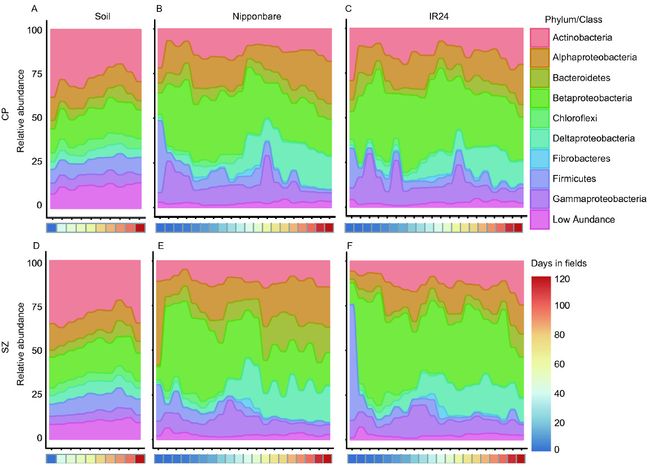
图3. 水稻根系微生物组主要菌门随时间变化(其中变形菌门丰度过高,进一步分为四个主要的纲)。图中6个子图分别展示了两个地点的土壤及两个品种的微生物组随时间规律的变化。
方法简介:此图主要使用R包ggaluvial绘制冲击图,时间轴采用pheatmap绘制热图(详见2a-相关分析)。
绘图实战
统计分析,主要基于两个表:OTU表和实验设计表,对于想进一步讨论分类级,别需要OTUs的物种注释文件。
这样基于这3个文件,可以制作出千变万化的统计分析图片,来作为论据支持你的文章(Story)。
清空工作环境和加载包
## Basic plotting stuff
# Set working enviroment in Rstudio, select Session - Set working directory - To source file location, default is runing directory
rm(list=ls()) # clean enviroment object
library("reshape2", quietly=T, warn.conflicts=F)
library(ggalluvial)
# Set ggplot2 drawing parameter, such as axis line and text size, lengend and title size, and so on.
main_theme = theme(panel.background=element_blank(),
panel.grid=element_blank(),
axis.line.x=element_line(size=.5, colour="black"),
axis.line.y=element_line(size=.5, colour="black"),
axis.ticks=element_line(color="black"),
axis.text=element_text(color="black", size=7),
legend.position="right",
legend.background=element_blank(),
legend.key=element_blank(),
legend.text= element_text(size=7),
text=element_text(family="sans", size=7))
读入实验设计、OTU表和物种注释
# Public file 1. "design.txt" Design of experiment
design = read.table("../data/design.txt", header=T, row.names= 1, sep="\t")
# Public file 2. "otu_table.txt" raw reads count of each OTU in each sample
otu_table = read.delim("../data/otu_table.txt", row.names= 1, header=T, sep="\t")
# Public file 3. "rep_seqs_tax.txt" taxonomy for each OTU, tab seperated
taxonomy = read.delim("../data/rep_seqs_tax.txt", row.names= 1,header=F, sep="\t")
colnames(taxonomy) = c("kingdom","phylum","class","order","family","genus","species","evalue")
# 物种注释只有门、纲、目等,而植物富集的Proteobacteria非常多,常是把其进一步分类为纲
# 获取均值topN的taxonomy+class信息,并分类汇总
# select p__Proteobacteria line
idx = taxonomy$phylum == "p__Proteobacteria"
# 初始化full为门,并初化因子为字符方便修改
taxonomy$full=as.character(taxonomy$phylum)
# 修改Proteobacteria门为目
taxonomy[idx,]$full=as.character(taxonomy[idx,]$class)
# 追加物种注释
tax_count = merge(taxonomy, otu_table, by="row.names")
# 按第10列门+纲组合,对OTU表进行分类汇总
tax_count_sum = aggregate(tax_count[,-(1:10)], by=tax_count[10], FUN=sum) # mean
# 汇总后需重新添加行名
rownames(tax_count_sum) = tax_count_sum$full
# 删除汇总列,变为纯数值矩阵
tax_count_sum = tax_count_sum[,-1]
# 标准化原始reads count为百分比
per = t(t(tax_count_sum)/colSums(tax_count_sum,na=T)) * 100 # normalization to total 100
筛选高丰度门用于展示
# 绘制样品组内各样品堆叠图
# 需要合并低丰度门,控制物种分类10种以内颜色展示才可识别
# 按丰度排序,目前门+变形菌纲有69类
mean_sort = per[(order(-rowSums(per))), ] # decrease sort
colSums(mean_sort)
# 筛选前9大类,其它归为Low abundance;只剩10组展示提高可读性
mean_sort=as.data.frame(mean_sort)
other = colSums(mean_sort[10:dim(mean_sort)[1], ])
mean_sort = mean_sort[1:(10-1), ]
mean_sort = rbind(mean_sort,other)
rownames(mean_sort)[10] = c("Low Abundance")
# 可视化前的表格保存,方便以后继续从这里开始使用
write.table(mean_sort, file="Top10phylum_ProClass.txt", append = F, sep="\t", quote=F, row.names=T, col.names=T)
# 保存高丰度列表
topN=rownames(mean_sort)
数据交叉筛选
图中有6个子图,我们以E图为例进行绘制,即上庄地点(Sz)种植的日本晴Nippobare(A50)时间序列样品进行统计
# 手动筛选实验中子集组
sub_design = subset(design,groupID %in% c("A50Sz0","A50Sz1","A50Sz2","A50Sz3","A50Sz5","A50Sz7","A50Sz10","A50Sz13","A50Sz27","A50Sz34","A50Sz41","A50Sz48","A50Sz56","A50Sz62","A50Sz69","A50Sz76","A50Sz83","A50Sz90","A50Sz97","A50Sz118") )
# 统计分组名为group
sub_design$group=sub_design$groupID
# 设置显示顺序,否则按字母排序(11会排在1后面)
sub_design$group = factor(sub_design$group, levels=c("A50Sz0","A50Sz1","A50Sz2","A50Sz3","A50Sz5","A50Sz7","A50Sz10","A50Sz13","A50Sz27","A50Sz34","A50Sz41","A50Sz48","A50Sz56","A50Sz62","A50Sz69","A50Sz76","A50Sz83","A50Sz90","A50Sz97","A50Sz118"))
# 统计本次分析的组数
print(paste("Number of group: ",length(unique(sub_design$group)),sep="")) # show group numbers
# 实验设计与物种相关丰度表交叉筛选
idx = rownames(sub_design) %in% colnames(mean_sort)
sub_design = sub_design[idx,]
mean_sort = mean_sort[, rownames(sub_design)] # reorder according to design
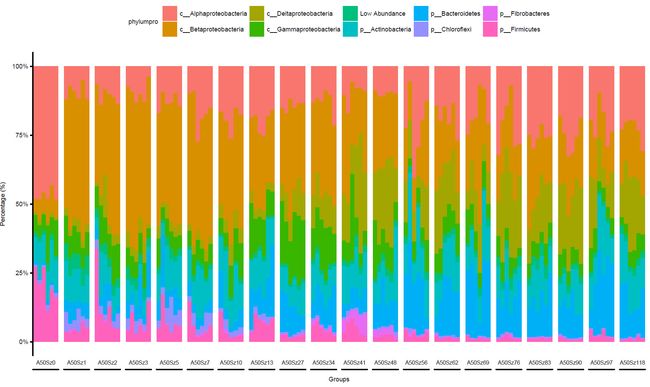
按样品绘图
# 添加图中物种分组
mean_sort$phylumpro = rownames(mean_sort)
# 矩阵表格转换为索引表格
data_all = as.data.frame(melt(mean_sort, id.vars=c("phylumpro")))
# 添加分组信息
data_all = merge(data_all, sub_design[c("group")], by.x="variable", by.y = "row.names")
# 按样品绘图
p = ggplot(data_all, aes(x=variable, y = value, fill = phylumpro )) +
geom_bar(stat = "identity",position="fill", width=1)+
scale_y_continuous(labels = scales::percent) +
# 分面,进一步按group分组,x轴范围自由否则位置异常,swith设置标签底部,并调置去除图例背景
facet_grid( ~ group, scales = "free_x", switch = "x") + main_theme +
# 关闭x轴刻度和标签
theme(axis.ticks.x = element_blank(), legend.position="top", axis.text.x = element_blank(), strip.background = element_blank())+
xlab("Groups")+ylab("Percentage (%)")
p
ggsave("tax_stack_phylumpro_sample.pdf", p, width = 10, height = 6)
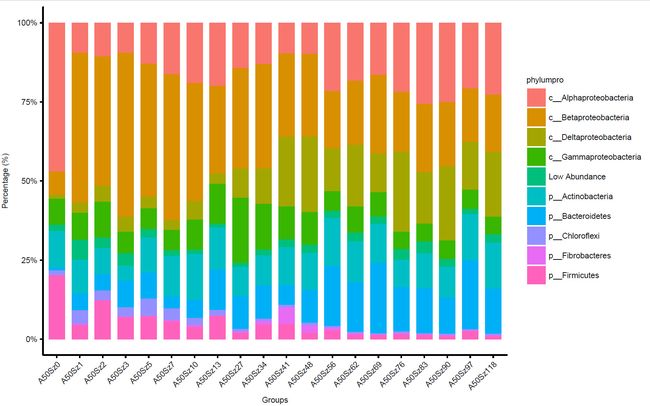
按组绘图
需要样品按组合并为均值
# 去除刚才添加的物种列
mat = mean_sort[,1:(dim(mean_sort)[2]-1)]
# 转换后样本为行名
mat_t = t(mat)
# 按行追加组名
mat_t2 = merge(sub_design[c("group")], mat_t, by="row.names")
# 删除合并后多余列
mat_t2 = mat_t2[,-1]
# 分组求均值
mat_mean = aggregate(mat_t2[,-1], by=mat_t2[1], FUN=mean) # mean
mat_mean_final = do.call(rbind, mat_mean)[-1,]
# 恢复行名
geno = mat_mean$group
colnames(mat_mean_final) = geno
# 转换为数据框
mat_mean_final = as.data.frame(mat_mean_final)
# 添加物种列
mat_mean_final$phylumpro = rownames(mat_mean_final)
# 表格转换
data_all = as.data.frame(melt(mat_mean_final, id.vars=c("phylumpro")))
p = ggplot(data_all, aes(x=variable, y = value, fill = phylumpro )) +
geom_bar(stat = "identity",position="fill", width=0.7)+
scale_y_continuous(labels = scales::percent) +
xlab("Groups")+ylab("Percentage (%)")+main_theme+ theme(axis.text.x = element_text(angle = 45, hjust = 1))
p
ggsave("tax_stack_phylumpro_top9.pdf", p, width = 8, height = 5)
绘制冲击图alluvium
p = ggplot(data = data_all, aes(x = variable, weight = value, alluvium = phylumpro)) +
geom_alluvium(aes(fill = phylumpro, colour = phylumpro, colour = phylumpro), alpha = .75) +
main_theme + theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
ggtitle("Phylum and class changes among groups")
p
ggsave("tax_alluvium_phylumpro.pdf", p, width = 8, height = 5)

本分析的全部文件和代码,会在 https://github.com/YongxinLiu/RiceTimeCourse 上持续更新,也可以后台回复“时间序列”获得百度云下载链接。
如果本文分享的技术帮助了你的科研,欢迎引用下文,支持国产SCI越来越好。
Citition:
Zhang, J., Zhang, N., Liu, Y.X., Zhang, X., Hu, B., Qin, Y., Xu, H., Wang, H., Guo, X., Qian, J., et al. (2018). Root microbiota shift in rice correlates
with resident time in the field and developmental stage. Sci China Life Sci 61, https://doi.org/10.1007/s11427-018-9284-4
点我下载PDF
猜你喜欢
- 10000+:肠道细菌 人体上的生命 宝宝与猫狗 梅毒狂想曲 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊
- 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板 Shell R Perl
- 生物科普 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外1700+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”

点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA