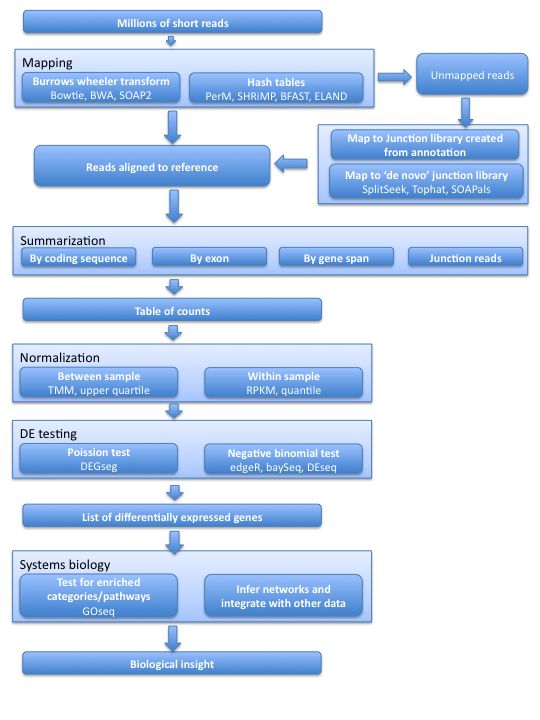
基因表达标准化Normalisation
我们在比较不同样品不同基因的差异表达情况时,期望表达水平分布符合统计方法的基本假设,但由于测序深度和基因长度的不同,直接使用原始count分析会导致假阳性和假阴性过高,因此对原始数据进行标准化/均一化是十分必要的。
根据样本间和样本内重复可以把现有的诸多标准化方法大致分为两类,一类WSN(within-sample normalization):RPKM和quartile四分位数法;另一类BSN(between-sample normalization):TMM和upper quartile上四分位处理。
WSN(within-sample normalization)
最普遍的做法是计算CPM (Counts Per Million),即原始reads count除以总reads数乘以1,000,000。CPM按照基因或转录本长度归一化后的表达即RPKM (Reads Counts Per Million)、FPKM (Fragments Per Kilobase Million)和TPM (Trans Per Million),推荐使用TPM(原理如图)。
calc_cpm <- function (expr_mat, spikes = NULL){
norm_factor <- colSums(expr_mat[-spikes,])
return(t(t(expr_mat)/norm_factor)) * 10^6
}


BSN(between-sample normalization)
为了解决上述问题,BSM类分出control set去评估测序深度而不是用所有数据,主要分三种:
TMM (trimmed mean of M-values)
TMM是M-值的加权截尾均值,即选定一个样品为参照,其它样品中基因的表达相对于参照样品中对应基因表达倍数的log2值定义为M-值。随后去除M-值中最高和最低的30%,剩下的M值计算加权平均值,权重来自Binomial data的delta方法 (Robinson and Oshlack, 2010)。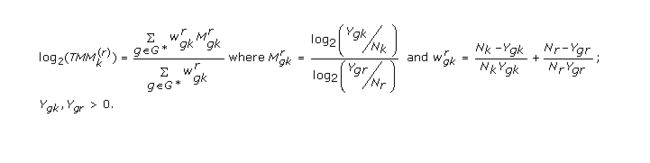
RLE (relative log expression)
RLE (relative log expression) 首先计算每个基因在所有样品中表达的几何平均值。然后再计算该值与每个样品的比值的中位数,也叫被称为量化因子scale factor (Anders and Huber 2010)。
calc_sf <- function (expr_mat, spikes=NULL){
geomeans <- exp(rowMeans(log(expr_mat[-spikes,])))
SF <- function(cnts){
median((cnts/geomeans)[(is.finite(geomeans) & geomeans >0)])
}
norm_factor <- apply(expr_mat[-spikes,],2,SF)
return(t(t(expr_mat)/norm_factor))
}
UQ (upper quartile)
上四分位数 (upper quartile, UQ)是样品中所有基因的表达除以处于上四分位数的基因的表达值。同时为了保证表达水平的相对稳定,计算得到的上四分位数值要除以所有样品中上四分位数值的中位数。
calc_uq <- function (expr_mat, spikes=NULL){
UQ <- function(x) {
quantile(x[x>0],0.75)
}
uq <- unlist(apply(expr_mat[-spikes,],2,UQ))
norm_factor <- uq/median(uq)
return(t(t(expr_mat)/norm_factor))
}
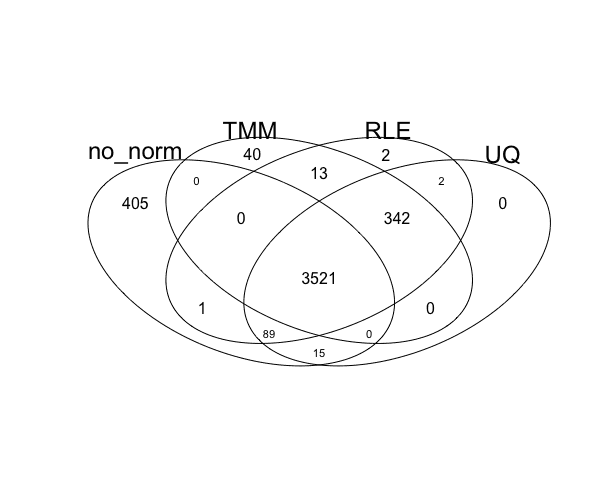
以上三种方法效果大同小异,通常比较流行的是TMM和DESeq normalization
edgeR中三种标准化TMM\UQ\RLE方法的比较以及不做标准化的后果
WSN和BSN两类方法的选择
没有定论,这方面研究较少,也可以先用BSN处理counts再做一下WSN。“Every normalization technique that I have seen assumes you are modeling counts, so the assumptions might be violated if you are using them directly on TPM or FPKM. While this is true, I think most techniques will give reasonable results in practice. Another possibility is to apply a BSN technique to the counts, then perform your within-sample normalization. This area has not been studied well, though we are actively working on it.”
From blog In RNA-Seq, 2 != 2: Between-sample normalization (里面有很多资料文献,可以进一步了解)
强烈推荐:
基因课视频
转录组原理篇
推文
RNA-Seq分析|RPKM, FPKM, TPM, 傻傻分不清楚?
Normalisation methods implemented in edgeR
DESeq2差异基因分析和批次效应移除
RNA-seq中的基因表达量计算和表达差异分析
What the FPKM? A review of RNA-Seq expression units
P.S. 这些天看着果子哥在微信号里笔耕不辍,感觉自己要更努力。特别是他最近发布的针对RNA-seq reads count批次效应处理的内容,我有些困惑,与其等着他人解答,不如自己主动找答案。正好那篇推文的英文参考链接里有详细的说明,了解到edgeR里三种均一化normalisation方法TMM,UQ和RLE的异同,而批次效应应该是要单独考虑的,今天先讲讲均一化,帮助自己能更新相关的基础概念,优化自己RNA-seq数据处理的流程,常学常新
果子学生信
理解 Quntile Normalization
批次效应这样矫正