转录组入门3:了解fastq测序数据
目的:用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量。
作业:理解测序reads,GC含量,质量值,接头,index,fastqc的全部报告,搜索中文教程
一. SRA Toolkit官方文档
https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc
Frequently Used Tools:
fastq-dump: Convert SRA data into fastq format
prefetch: Allows command-line downloading of SRA, dbGaP, and ADSP data
sam-dump: Convert SRA data to sam format
sra-pileup: Generate pileup statistics on aligned SRA data
vdb-config: Display and modify VDB configuration information
vdb-decrypt: Decrypt non-SRA dbGaP data ("phenotype data")
Additional Tools:
abi-dump: Convert SRA data into ABI format (csfasta / qual)
illumina-dump: Convert SRA data into Illumina native formats (qseq, etc.)
sff-dump: Convert SRA data to sff format
sra-stat: Generate statistics about SRA data (quality distribution, etc.)
vdb-dump: Output the native VDB format of SRA data.
vdb-encrypt: Encrypt non-SRA dbGaP data ("phenotype data")
vdb-validate: Validate the integrity of downloaded SRA data
SRA Toolkit Installation and Configuration Guide
Testing the Toolkit configuration
download repository(Linux): /home/[user_name]/ncbi/public
For the test, we are using an arbitrary dataset, SRR390728 (RNA-Seq (polyA+) analysis of DLBCL cell line HS0798), from the National Cancer Institute’s Cancer Genome Characterization Initiative (CGCI) Project. It is a reasonably small SRA dataset that contains aligned (reference-compressed) data, allowing us to test multiple aspects of the toolkit simultaneously.
- Open a terminal or command prompt and “cd” into the directory containing the toolkit executables (e.g., [download_location]/sratoolkit[version]/bin/).
Linux and OS X users should execute the following command:
./fastq-dump -X 5 -Z SRR390728
- If successful, the test should connect to NCBI, download a small amount of data from SRR390728 and the reference sequence needed to extract the data, and stream the first 5 spots of the file ("-X 5" option) to the screen ("-Z" option).
- If the configuration is not valid, an error like the following will likely be displayed:
fastq-dump.2.x err: item not found while constructing within virtual database module - the path 'SRR390728' cannot be opened as database or table"
- If you receive an error like the one above, please configure the toolkit (described in the next section). If you have already configured the toolkit but are still unable to complete the test successfully, please email [email protected] with a full description of steps taken and error messages received.
Configuring the Toolkit
Go to the “bin” subdirectory for the Toolkit and run the following command:
./vdb-config -i
本地文档
fastq-dump -h #显示帮助
Usage:
fastq-dump [options] <path> [<path>...]
fastq-dump [options] <accession>
INPUT
-A|--accession <accession> Replaces accession derived from <path> in
filename(s) and deflines (only for single
table dump)
--table <table-name> Table name within cSRA object, default is
"SEQUENCE"
OUTPUT
-O|--outdir <path> Output directory, default is working
directory '.' )
-Z|--stdout Output to stdout, all split data become
joined into single stream
--gzip Compress output using gzip: deprecated, not
recommended
--bzip2 Compress output using bzip2: deprecated,
not recommended
Multiple File Options Setting these options will produce more
than 1 file, each of which will be suffixed
according to splitting criteria.
FASTQ文件每个序列通常为4行,分别为:
Line 1 begins with a ‘@’ character and is followed by a sequence identifier and an optional description (like a FASTA title line).
Line 2 is the raw sequence letters.
Line 3 begins with a ‘+’ character and is optionally followed by the same sequence identifier (and any description) again.
Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
转换及检测测序质量
sra转换为fastq
fastq-dump --split-3 -O SRR35899$i.sra
# 翻车了,不压缩有120G,推荐压缩 --gzip
# 利用循环减少重复操作
检测质量
fastqc SRR3589956_1.fastq
得到一个zip压缩文件和一个html文件
打开html文件获得检测结果

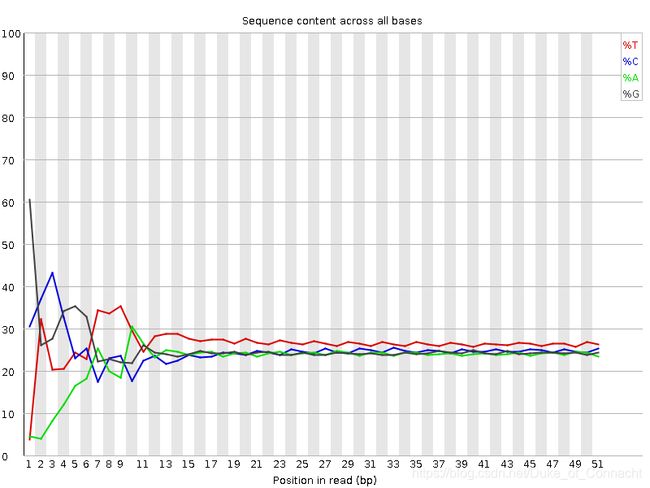
使用MultiQC检测质量
青山屋主专栏:
http://fbb84b26.wiz03.com/share/s/3XK4IC0cm4CL22pU
r1HPcQQ1iRTvV2GwkwL2AaxYi2fXHP7
conda install -c bioconda multiqc
multiqc . # 扫描当前文件夹

由图可知检测质量理想
感谢生物技能树论坛,本笔记根据各位朋友提供的笔记与学习材料完成