- 今日记事--餐厅记事
星辰大海2021
工作日的每天中午,我都去餐厅吃饭。先从实验室出来,下楼,骑上我的凤凰自行车,向南,路过图书馆,拐个弯就到了东苑餐厅,锁车,进电梯,直上3楼,下电梯,左拐,走到楼梯口,那盘子,转身,拿筷子。筷笼对面就是选餐区。一盆盆菜摆放好了,供您挑选。一般先是热菜,大约有6盆,经常常见的有回锅肉,宫爆鸡丁,蒜苔炒肉,豆角炒肉,土豆鸡块,辣椒炒肉,西红柿炒鸡蛋,豆腐块炒肉。今天多了俩油炸食品,一个是土豆块,我才开始
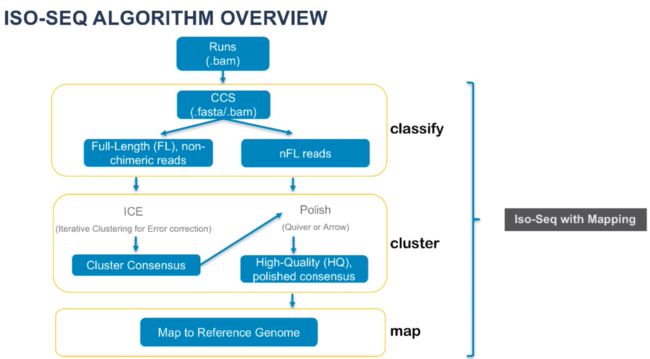
- 【算法-贪心算法-python】柠檬水找零
檀越@新空间
P1算法与数据结构s1Python算法贪心算法python
欢迎来到我的博客,很高兴能够在这里和您见面!希望您在这里可以感受到一份轻松愉快的氛围,不仅可以获得有趣的内容和知识,也可以畅所欲言、分享您的想法和见解。推荐:kuan的首页,持续学习,不断总结,共同进步,活到老学到老导航檀越剑指大厂系列:全面总结java核心技术点,如集合,jvm,并发编程redis,kafka,Spring,微服务,Netty等常用开发工具系列:罗列常用的开发工具,如IDEA,M
- 经常安慰自己的一句话是什么?
余子烨
活着就好,健健康康的活着更好了,开开心心的活着就最好了。或许是经历的事情太多了,或许是生活太过于艰难了。有时候真的感觉活着就好,当身边有人患了重病癌症而离开人世,当看到疫情中有人失去生病,当自己亲人生病进了医院,就深切的感觉活着就好,健健康康的活着就更好了,如果再大的追求就是开开心心的活着。人生的追求不就是开开心心的活着吗?人生的幸福不就是开开心心的活着吗?人活着一辈子就是为了好好地,开开心心地活
- uniapp写好的弹窗组件
A了LONE
uni-app前端
效果图view部分点击打开弹窗确认退款是否确认申请退款?取消确定js部分data(){return{miniShowModal:false,//默认隐藏弹框}},methods:{//点击按钮弹出弹框miniToMdel(){this.miniShowModal=true;},//点击确定按钮时关闭弹框confirm(){this.closeOn()},//点击蒙版时关闭按钮miniHideMod
- uniapp--腾讯地图路线轨迹回放
前端志茗
uni-appjson前端微信小程序小程序
腾讯地图路线轨迹回放返回路线轨迹手动选择目的地开始驾车路线规划显示小车轨迹模拟运行//引入SDK核心类,地图组件importQQMapWXfrom'../components/qqmap-wx-jssdk1.2/qqmap-wx-jssdk.js'exportdefault{data(){return{qqmapsdk:{},//腾讯地图小程序的SDKtext:'路线轨迹,带小车图标',//滚动通
- exports使用 package.json字段控制如何访问你的 npm 包
前端 贾公子
jsonnpm前端
目录想象一下你正在开发一个npm包……术语什么是exports领域?exports好处保护内部文件多格式包将子路径映射到dist目录子路径导出单一入口点多个入口点公开软件包文件的子集有条件出口设置使用条件默认条件句法针对Node.js和浏览器想象一下你正在开发一个npm包……您希望提供多个入口点,但同时限制对内部文件的访问。您需要同时支持CJS和ESM,包含类型定义,甚至可能还要确保浏览器兼容性。
- 由己及彼
和佛陀去赏花
王冬冬,中原焦点团队讲师、心理咨询师,持续记录987天(2020.8.22)雨农历七月初三,庚子鼠年甲申月丁酉日,处暑第一候鹰乃祭鸟,末伏第八天。阅读打卡第777天:《老子》《漫步中国星空》《五运六气》《焦点解决治疗理论研究与实践》朗诵记录第778天:诵读第112周第6天,《老子》一整天的忙碌,都没顾上想念泉哥。晚上接到泉哥,迫不及待的询问在校情况,吃的如何?交到几个朋友等。泉哥兴奋的说:没想到学
- 人生下半场,学会放过自己
是小萌姐姐呀
01.听过这么一句话:与其在别人的目光中消逝自我,不如在黑暗中闪耀自己。心理学上有一个名词:聚光灯效应,也称作焦点效应。讲的是太在乎和自己有关的事情,总以为别人的目光都聚集在自己的身上。如果每做一件事情,总是考虑别人的感觉怎样,那便失去了对自己衡量的标准。大多数人的定力和毅力并没有强大到对任何事情都可以安然处之。在意别人的目光,并没有什么错,可是过分在意就只会把自己累得够呛。不要把别人的标准当成自
- 机会都是给有准备的人的
岩龙的书
前几天预约了牙科医生,好不容易请到假,提早两天就关注天气,预报说是有雷暴雨奥我的天,没想昨儿个好朋友又来了,啊我的妈真的有种屋漏偏逢连夜雨的赶脚,什么事都碰到一起来,昨天担心了一天,后来就想算了,还是去吧,该来的就来,能咋滴,往后推迟只会延迟时间,打定主意就不想了,疼也去,下刀子也去。本来预约的时间要很晚,担心接小小只晚点,就早走一小时,按理来说预约几点就是几点看的,本想在外消磨掉那一小时在去医院
- 2023-7-27晨间日记
辽远的边疆
今天是什么日子起床:就寝:天气:心情:纪念日:任务清单昨日完成的任务,最重要的三件事:改进:习惯养成:周目标·完成进度学习·信息·阅读健康·饮食·锻炼人际·家人·朋友工作·思考最美好的三件事1.2.3.思考·创意·未来
- 贵妃替身将我千刀万剐后,疯批暴君杀疯了(凌晟舒纪雨)全文免费阅读_最热门小说 贵妃替身将我千刀万剐后,疯批暴君杀疯了凌晟舒纪雨
云朵美文
《贵妃替身将我千刀万剐后,疯批暴君杀疯了》主角:凌晟舒纪雨简介:我是疯批暴君求而不得的白月光。他曾下令荒废六宫,许我专宠,只为求我一笑。可我不爱,以死拒绝。暴君怒了,将我囚禁在深宫之中。他又纳了无数与我容貌相似的女子,进后宫为妃。不到三天,便要再换新宠。但我听说,这一回他对一位女子椒房专宠,甚至还晋升为贵妃。贵妃认为自己定是后宫之主,闯入幽宫,说要替皇帝严惩狐媚子。见到我那一刻,她妒火中烧:“就凭
- 伪数组转换为真正的数组
会飞的鱼先生
javascript前端vue.js
在JavaScript中,**伪数组(类数组对象)**是指具有数字索引和length属性,但不具备数组原生方法的对象。常见的伪数组包括函数的arguments对象、DOM集合(如document.querySelectorAll的返回值)等。要将伪数组转换为真正的数组,可以使用以下几种方法:1.使用Array.from()Array.from()是一种简洁且高效的方法,可以将伪数组转换为真正的数组
- stable diffusion-系统课程:0基础系统性学习AI绘画,小白也能轻松上手
顺心网创
本课程是AI绘画工具stablediffusion的系统课程,内容通俗且细致,让小白也能上手。课程大纲基础部分1.前置要求+整合包安装+启动器使用2.纯净原版安装+使用介绍3.文生图精讲4.图生图精讲5.涂鸦、局部重绘、涂鸦重绘6.上传蒙版、批量处理7.模型精讲8.提示词精讲9.插件的认识与安装10.脚本的安装及使用11controlnet基础讲解12.cn-线性控制类型13.cn-深度和法线进阶
- Ubuntu安装Electron环境
咖喱鱼蛋
Web项目记录ubuntuelectronlinux
前言Electron官方文档要开发Electron应用,您需要安装Node.js运行环境和它的包管理器npm。我们推荐安装最新的长期支持(LTS)版本。 安装nvmnode.js的版本管理工具curl-o-https://raw.githubusercontent.com/nvm-sh/nvm/v0.39.1/install.sh|bash 安装最新的LTS版本的Node.jsnvminsta
- 适合宝妈在家做的副业赚钱方法有哪些?盘点宝妈在家可做的六种赚钱方法
高省APP大九
作为宝妈,您可能需要在照顾家庭和孩子的同时,也想要找到一种方式增加家庭收入。随着互联网的普及,越来越多的宝妈选择在家做副业来实现这一目标。本文将为您介绍几种适合宝妈在家做的副业赚钱方法,帮助您找到适合自己的赚钱途径。一、电商推广与社交分享电商推广与社交分享是另一种适合宝妈在家做的副业赚钱方式。您可以利用自己的社交圈,通过微信、微博等社交平台分享优质商品链接或优惠券,引导朋友和家人购买。一旦他们通过
- 星际:追我的全是大佬精品小说在线观看(秋凌、苍昔)最新章节无弹窗广告_星际:追我的全是大佬精品全文阅读
霸道推书3
小说简介:飞升失败后,我决定摆烂了,男人什么的直接听从系统安排,它给我啥男人,我照单全收算了。但是,系统这小子选的男人可真对我胃口啊,那我还是拾掇拾掇自己,拿下这些男人的心吧。书名:《星际:追我的全是大佬》主角配角:秋凌、苍昔推荐指数:✩✩✩✩✩———小说内容试读———颜诏点头,特别贤惠地说:“好,我正好在网上看看食堂里还有什么饭,让人送过来。等你忙完了,我们—起吃。”秋凌想起他刚才说的,替她张罗
- js数组与字符串方法
拼接方法字符串拼接用+就行了,如str1+str2。数组则用join()方法把元素连起来,比如arr.join(‘,’)。数组不能直接用+拼接,但两个数组相加会被转成字符串再拼接,这不是数组的拼接,而是字符串拼接行为。letstr1="Hello";letstr2="World";letarr=['Hello','World'];console.log(str1+""+str2);//"Hello
- 老师晨说
平安413
主题【驯化我们的大脑】️驯化脑神经回路练习临在当下,觉察大脑神经元变化-过去:思前想后犹犹豫豫,内心戏不断加码,担忧未来放不下过去-现在:什么都不想,没有任何担忧,活在当下感知当下。现在大脑的运作模式已经完全不对什么事产生应激反应。️重视刻意训练坚定信念、信仰——训练可以带你进入幸福和可能性。训练出对当下的全心全意,全然投入在当下事情。训练对幸福感知回路,当下全心全意来到美好当中。最终把自己训练成
- 最大高仿服装市场,3分钟讲解攻略及经验
天坛众
中国最大的高仿服装市场有几个比较典型的代表,分别是:拿货微信:377267298(手表、鞋子包包服装首饰皮带等)1.义乌中国小商品城:位于浙江省义乌市,是世界上最大的小商品市场之一,其中包括了大量的服装产品。2.广州白马服装市场:位于广州市,是全国著名的女装批发市场,以高品质和中高端市场定位著称。3.杭州四季青服装市场:位于杭州市,是中国最大的服装批发市场之一,以女装批发为主。4.即墨服装市场:位
- 【用unity实现100个游戏之34】使用环状(车轮)碰撞器(Wheel Collider)从零实现一个汽车车辆物理控制系统,实现一个赛车游戏
向宇it
【制作100个Unity游戏】unity游戏汽车游戏引擎3d材质
最终效果unity赛车效果文章目录最终效果前言一、WheelCollider参数介绍1、基础参数2、SuspensionSpring:悬挂弹簧2.1spring支撑悬挂的弹力2.3damper减震2.4targetposition:表示车轮静止时处于的悬挂上的位置3、forwardfriction前向摩檫力和sidewaysfriction侧向摩檫力二、准备工作1、下载素材2、给车辆添加Rigid
- 播音丨纪录片配音到底要注意啥?
播音配音学习攻略
纪录片像是一个记录真实生活,或真人真事的艺术展现形式,在众人的眼中一直是以真实为主。而纪录片的配音也会根据不同的素材类型稍作更改,但整体来说,纪录片的配音大多数是以辅助的形式将人、事、物,清楚的表达在大家的视野中~不过纪录片配音时,到底应该注意什么呢?想必还是很多人不太了解,今天就来分享两点比较基础,但是又不得不注意的吧~把握好音量及用声电视纪录片的用声,大多音量会比广播配音要小,这种解说式的配音
- 2023-09-09
b2a585406465
“回报家乡人民——李荣海捐赠菏泽市人民政府书画作品六百幅大型回报展”作品欣赏(第五期)菏泽市李荣海美术馆菏泽市艺术博物馆7月26日,“回报家乡人民——李荣海捐赠菏泽市人民政府书画作品大型回报展”在菏泽市李荣海美术馆(菏泽市艺术博物馆)拉开帷幕,这是继李荣海先生捐赠美术馆后的又一义举,对推动菏泽文化建设具有重要意义。本次展览得到菏泽市领导、艺术界人士、院校师生、新闻媒体的广泛关注和高度评价。李荣海先
- 别人发来的消息,你会及时回吗?
雨果的天空
现在是信息爆炸的时代,每天接触的信息太多了,朋友的消息、️群里的互动、朋友圈的动态铺天盖地,纷纷扰扰好不热闹。面对别人发来的消息,你会及时回复吗?我觉得首先要看对象是谁?如果是重要的领导、挚爱的亲人或自己在意的人,我都会及时回复。其实冷静下来细思量,给你发消息的也有三六九等,对方在你心里的位置,会决定你对TA的态度。我始终认为,回信息也是人起码的礼貌。学着换位思考一下,万一自己的消息发给特别在意的
- 2019-01-19
常庚_c041
姓名:林常庚(365期,乐观3组)单位:深圳市蔚蓝时代商业管理有限公司海南分公司。【日精进打卡第289天】【知~学习《六项精进》1遍共298遍《大学》0遍共284遍《六项精进通篇》0遍共9遍【经典名句人生是主管世界与客观世界之间的那道沟,掉进去叫失败,爬出来就叫做成功人不容我是我无能,我不容人是我量【行~实践一、修身:(对自己二、齐家:(对家庭和家给妈妈买年货三、建功:(对工作休假积善:发愿从20
- 大单元教学反思之三
248广州刘在丽
2.没能兼顾学生教辅作业上的部分题目,像修辞手法的判断、课外文章的理解与分析等题目,一直带上来的班级学生做题稍微能结合模糊的旧知识答题,新接班的基础薄弱的学生基本上属于乱做,连修辞手法的判断都没有积累到方法。只好在讲解的时候再次渗透方法。3.学生的书写也容易忽略。因为上生字词基本上一课时完成,既要抓过关又要抓重难点生字的笔画笔顺间架结构,感觉时间不够用。只能依靠学生一二年级积累的书写经验和对写作业
- 国庆套优惠券怎么获得?国庆节购物优惠卷的获取技巧
高省APP珊珊
国庆套优惠券的获取方式及国庆节购物优惠券的获取技巧,主要依赖于具体的商品或服务类型以及对应的商家促销活动。以下是一些通用的获取技巧和建议:国庆套优惠券获取方式(以游戏为例)以游戏(如DNF)中的国庆套优惠券为例,通常有以下几种获取方式:购买前置礼包:部分游戏会设置前置礼包,玩家在购买这些礼包后,可以获得国庆套优惠券作为额外奖励。例如,在DNF中,购买夏日海上礼包就可能获得国庆套优惠券。参与游戏内活
- 操场上的欢乐
Wyf成长瞬间
今天中午吃饱饭以后,我们和好朋友约定好到学校旁边的操场上踢球。我们带着足球和小车来到操场上,里面的人非常多,到处一片欢声笑语,我们所有的队员分成两组,一组是守门员,另一组负责进攻射球,我们用力的用脚把球踢向球门,守门员接住了,我们就输了,如果守门员没接住,我们就赢了,我们玩的不亦乐乎,战斗非常的激烈。时间过得很快,已经到了回家的时间了,我们只好依依不舍的和小伙伴们说了一声再见之后,就离开啦!今天我
- 彭放感悟《论语》的人文情怀之【9.13】——待价而沽不藏于匵
教育参悟人
【9.13】子贡曰:“有美玉于斯,韫椟而藏诸?求善贾而沽(卖出去)诸?”子曰:“沽之哉,沽之哉!我待贾者也。”子贡说:“这里有一块美玉,是把它收藏在匣子里呢?还是找一个识货的商人卖出去呢?”孔子说:“卖掉吧,卖掉吧!我正在等待识货的人呢。”【感悟】老师有许多治国之道,却英雄无用武之处,子贡巧设譬喻。孔子自称是“待贾者”,他一方面四处游说,以宣传礼治天下为己任,期待着各国统治者能够行他之道于天下;另
- 【gateway网关】
叫我李老板
gateway学习php
网关的核心功能网关(Gateway)作为网络架构中的关键组件,主要承担不同协议或网络之间的数据转换与路由功能。以下是其核心功能的详细说明:协议转换与适配网关能够连接使用不同通信协议的网络或系统,实现数据格式的转换。例如将HTTP请求转换为gRPC协议,或处理SOAP与RESTfulAPI之间的互操作。这种能力在混合云环境或遗留系统集成中尤为重要。流量路由与负载均衡基于请求内容(如URL路径、HTT
- Linux系统修改时区以及校准时间
Linux系统修改时区以及校准时间修改时区(切换到root用户下执行suroot)删除系统自带的loacltime的文件rm-f/etc/loacltime将系统内置的时区文件Shanghai软连接到/etc/localtime(建议直接复制执行)ln-s/usr/share/zoneinfo/Asia/Shanghai/etc/localtime这个时候,就已经修改成功了,可以执行date看一下
- apache 安装linux windows
墙头上一根草
apacheinuxwindows
linux安装Apache 有两种方式一种是手动安装通过二进制的文件进行安装,另外一种就是通过yum 安装,此中安装方式,需要物理机联网。以下分别介绍两种的安装方式
通过二进制文件安装Apache需要的软件有apr,apr-util,pcre
1,安装 apr 下载地址:htt
- fill_parent、wrap_content和match_parent的区别
Cb123456
match_parentfill_parent
fill_parent、wrap_content和match_parent的区别:
1)fill_parent
设置一个构件的布局为fill_parent将强制性地使构件扩展,以填充布局单元内尽可能多的空间。这跟Windows控件的dockstyle属性大体一致。设置一个顶部布局或控件为fill_parent将强制性让它布满整个屏幕。
2) wrap_conte
- 网页自适应设计
天子之骄
htmlcss响应式设计页面自适应
网页自适应设计
网页对浏览器窗口的自适应支持变得越来越重要了。自适应响应设计更是异常火爆。再加上移动端的崛起,更是如日中天。以前为了适应不同屏幕分布率和浏览器窗口的扩大和缩小,需要设计几套css样式,用js脚本判断窗口大小,选择加载。结构臃肿,加载负担较大。现笔者经过一定时间的学习,有所心得,故分享于此,加强交流,共同进步。同时希望对大家有所
- [sql server] 分组取最大最小常用sql
一炮送你回车库
SQL Server
--分组取最大最小常用sql--测试环境if OBJECT_ID('tb') is not null drop table tb;gocreate table tb( col1 int, col2 int, Fcount int)insert into tbselect 11,20,1 union allselect 11,22,1 union allselect 1
- ImageIO写图片输出到硬盘
3213213333332132
javaimage
package awt;
import java.awt.Color;
import java.awt.Font;
import java.awt.Graphics;
import java.awt.image.BufferedImage;
import java.io.File;
import java.io.IOException;
import javax.imagei
- 自己的String动态数组
宝剑锋梅花香
java动态数组数组
数组还是好说,学过一两门编程语言的就知道,需要注意的是数组声明时需要把大小给它定下来,比如声明一个字符串类型的数组:String str[]=new String[10]; 但是问题就来了,每次都是大小确定的数组,我需要数组大小不固定随时变化怎么办呢? 动态数组就这样应运而生,龙哥给我们讲的是自己用代码写动态数组,并非用的ArrayList 看看字符
- pinyin4j工具类
darkranger
.net
pinyin4j工具类Java工具类 2010-04-24 00:47:00 阅读69 评论0 字号:大中小
引入pinyin4j-2.5.0.jar包:
pinyin4j是一个功能强悍的汉语拼音工具包,主要是从汉语获取各种格式和需求的拼音,功能强悍,下面看看如何使用pinyin4j。
本人以前用AscII编码提取工具,效果不理想,现在用pinyin4j简单实现了一个。功能还不是很完美,
- StarUML学习笔记----基本概念
aijuans
UML建模
介绍StarUML的基本概念,这些都是有效运用StarUML?所需要的。包括对模型、视图、图、项目、单元、方法、框架、模型块及其差异以及UML轮廓。
模型、视与图(Model, View and Diagram)
&
- Activiti最终总结
avords
Activiti id 工作流
1、流程定义ID:ProcessDefinitionId,当定义一个流程就会产生。
2、流程实例ID:ProcessInstanceId,当开始一个具体的流程时就会产生,也就是不同的流程实例ID可能有相同的流程定义ID。
3、TaskId,每一个userTask都会有一个Id这个是存在于流程实例上的。
4、TaskDefinitionKey和(ActivityImpl activityId
- 从省市区多重级联想到的,react和jquery的差别
bee1314
jqueryUIreact
在我们的前端项目里经常会用到级联的select,比如省市区这样。通常这种级联大多是动态的。比如先加载了省,点击省加载市,点击市加载区。然后数据通常ajax返回。如果没有数据则说明到了叶子节点。 针对这种场景,如果我们使用jquery来实现,要考虑很多的问题,数据部分,以及大量的dom操作。比如这个页面上显示了某个区,这时候我切换省,要把市重新初始化数据,然后区域的部分要从页面
- Eclipse快捷键大全
bijian1013
javaeclipse快捷键
Ctrl+1 快速修复(最经典的快捷键,就不用多说了)Ctrl+D: 删除当前行 Ctrl+Alt+↓ 复制当前行到下一行(复制增加)Ctrl+Alt+↑ 复制当前行到上一行(复制增加)Alt+↓ 当前行和下面一行交互位置(特别实用,可以省去先剪切,再粘贴了)Alt+↑ 当前行和上面一行交互位置(同上)Alt+← 前一个编辑的页面Alt+→ 下一个编辑的页面(当然是针对上面那条来说了)Alt+En
- js 笔记 函数
征客丶
JavaScript
一、函数的使用
1.1、定义函数变量
var vName = funcation(params){
}
1.2、函数的调用
函数变量的调用: vName(params);
函数定义时自发调用:(function(params){})(params);
1.3、函数中变量赋值
var a = 'a';
var ff
- 【Scala四】分析Spark源代码总结的Scala语法二
bit1129
scala
1. Some操作
在下面的代码中,使用了Some操作:if (self.partitioner == Some(partitioner)),那么Some(partitioner)表示什么含义?首先partitioner是方法combineByKey传入的变量,
Some的文档说明:
/** Class `Some[A]` represents existin
- java 匿名内部类
BlueSkator
java匿名内部类
组合优先于继承
Java的匿名类,就是提供了一个快捷方便的手段,令继承关系可以方便地变成组合关系
继承只有一个时候才能用,当你要求子类的实例可以替代父类实例的位置时才可以用继承。
在Java中内部类主要分为成员内部类、局部内部类、匿名内部类、静态内部类。
内部类不是很好理解,但说白了其实也就是一个类中还包含着另外一个类如同一个人是由大脑、肢体、器官等身体结果组成,而内部类相
- 盗版win装在MAC有害发热,苹果的东西不值得买,win应该不用
ljy325
游戏applewindowsXPOS
Mac mini 型号: MC270CH-A RMB:5,688
Apple 对windows的产品支持不好,有以下问题:
1.装完了xp,发现机身很热虽然没有运行任何程序!貌似显卡跑游戏发热一样,按照那样的发热量,那部机子损耗很大,使用寿命受到严重的影响!
2.反观安装了Mac os的展示机,发热量很小,运行了1天温度也没有那么高
&nbs
- 读《研磨设计模式》-代码笔记-生成器模式-Builder
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
/**
* 生成器模式的意图在于将一个复杂的构建与其表示相分离,使得同样的构建过程可以创建不同的表示(GoF)
* 个人理解:
* 构建一个复杂的对象,对于创建者(Builder)来说,一是要有数据来源(rawData),二是要返回构
- JIRA与SVN插件安装
chenyu19891124
SVNjira
JIRA安装好后提交代码并要显示在JIRA上,这得需要用SVN的插件才能看见开发人员提交的代码。
1.下载svn与jira插件安装包,解压后在安装包(atlassian-jira-subversion-plugin-0.10.1)
2.解压出来的包里下的lib文件夹下的jar拷贝到(C:\Program Files\Atlassian\JIRA 4.3.4\atlassian-jira\WEB
- 常用数学思想方法
comsci
工作
对于搞工程和技术的朋友来讲,在工作中常常遇到一些实际问题,而采用常规的思维方式无法很好的解决这些问题,那么这个时候我们就需要用数学语言和数学工具,而使用数学工具的前提却是用数学思想的方法来描述问题。。下面转帖几种常用的数学思想方法,仅供学习和参考
函数思想
把某一数学问题用函数表示出来,并且利用函数探究这个问题的一般规律。这是最基本、最常用的数学方法
- pl/sql集合类型
daizj
oracle集合typepl/sql
--集合类型
/*
单行单列的数据,使用标量变量
单行多列数据,使用记录
单列多行数据,使用集合(。。。)
*集合:类似于数组也就是。pl/sql集合类型包括索引表(pl/sql table)、嵌套表(Nested Table)、变长数组(VARRAY)等
*/
/*
--集合方法
&n
- [Ofbiz]ofbiz初用
dinguangx
电商ofbiz
从github下载最新的ofbiz(截止2015-7-13),从源码进行ofbiz的试用
1. 加载测试库
ofbiz内置derby,通过下面的命令初始化测试库
./ant load-demo (与load-seed有一些区别)
2. 启动内置tomcat
./ant start
或
./startofbiz.sh
或
java -jar ofbiz.jar
&
- 结构体中最后一个元素是长度为0的数组
dcj3sjt126com
cgcc
在Linux源代码中,有很多的结构体最后都定义了一个元素个数为0个的数组,如/usr/include/linux/if_pppox.h中有这样一个结构体: struct pppoe_tag { __u16 tag_type; __u16 tag_len; &n
- Linux cp 实现强行覆盖
dcj3sjt126com
linux
发现在Fedora 10 /ubutun 里面用cp -fr src dest,即使加了-f也是不能强行覆盖的,这时怎么回事的呢?一两个文件还好说,就输几个yes吧,但是要是n多文件怎么办,那还不输死人呢?下面提供三种解决办法。 方法一
我们输入alias命令,看看系统给cp起了一个什么别名。
[root@localhost ~]# aliasalias cp=’cp -i’a
- Memcached(一)、HelloWorld
frank1234
memcached
一、简介
高性能的架构离不开缓存,分布式缓存中的佼佼者当属memcached,它通过客户端将不同的key hash到不同的memcached服务器中,而获取的时候也到相同的服务器中获取,由于不需要做集群同步,也就省去了集群间同步的开销和延迟,所以它相对于ehcache等缓存来说能更好的支持分布式应用,具有更强的横向伸缩能力。
二、客户端
选择一个memcached客户端,我这里用的是memc
- Search in Rotated Sorted Array II
hcx2013
search
Follow up for "Search in Rotated Sorted Array":What if duplicates are allowed?
Would this affect the run-time complexity? How and why?
Write a function to determine if a given ta
- Spring4新特性——更好的Java泛型操作API
jinnianshilongnian
spring4generic type
Spring4新特性——泛型限定式依赖注入
Spring4新特性——核心容器的其他改进
Spring4新特性——Web开发的增强
Spring4新特性——集成Bean Validation 1.1(JSR-349)到SpringMVC
Spring4新特性——Groovy Bean定义DSL
Spring4新特性——更好的Java泛型操作API
Spring4新
- CentOS安装JDK
liuxingguome
centos
1、行卸载原来的:
[root@localhost opt]# rpm -qa | grep java
tzdata-java-2014g-1.el6.noarch
java-1.7.0-openjdk-1.7.0.65-2.5.1.2.el6_5.x86_64
java-1.6.0-openjdk-1.6.0.0-11.1.13.4.el6.x86_64
[root@localhost
- 二分搜索专题2-在有序二维数组中搜索一个元素
OpenMind
二维数组算法二分搜索
1,设二维数组p的每行每列都按照下标递增的顺序递增。
用数学语言描述如下:p满足
(1),对任意的x1,x2,y,如果x1<x2,则p(x1,y)<p(x2,y);
(2),对任意的x,y1,y2, 如果y1<y2,则p(x,y1)<p(x,y2);
2,问题:
给定满足1的数组p和一个整数k,求是否存在x0,y0使得p(x0,y0)=k?
3,算法分析:
(
- java 随机数 Math与Random
SaraWon
javaMathRandom
今天需要在程序中产生随机数,知道有两种方法可以使用,但是使用Math和Random的区别还不是特别清楚,看到一篇文章是关于的,觉得写的还挺不错的,原文地址是
http://www.oschina.net/question/157182_45274?sort=default&p=1#answers
产生1到10之间的随机数的两种实现方式:
//Math
Math.roun
- oracle创建表空间
tugn
oracle
create temporary tablespace TXSJ_TEMP
tempfile 'E:\Oracle\oradata\TXSJ_TEMP.dbf'
size 32m
autoextend on
next 32m maxsize 2048m
extent m
- 使用Java8实现自己的个性化搜索引擎
yangshangchuan
javasuperword搜索引擎java8全文检索
需要对249本软件著作实现句子级别全文检索,这些著作均为PDF文件,不使用现有的框架如lucene,自己实现的方法如下:
1、从PDF文件中提取文本,这里的重点是如何最大可能地还原文本。提取之后的文本,一个句子一行保存为文本文件。
2、将所有文本文件合并为一个单一的文本文件,这样,每一个句子就有一个唯一行号。
3、对每一行文本进行分词,建立倒排表,倒排表的格式为:词=包含该词的总行数N=行号