非靶向代谢组学数据分析方法总结
生物信息学早已不再局限于基因组学领域了,后基因组学越来越受到关注,并且这几年“多组学”的也研究越来越多。其中,代谢组学是相对比较年轻的一门学科,“代谢组”(metabolome)的概念于1998第一次被提出。基因组学和转录组学是生物信息的上游,更多的体现的是生物活动的内在本质因素,而代谢组学是生物信息的最下游,体现的是生物活动的表型结果。代谢组学分为靶向代谢组学和非靶向代谢组学,本文将结合本人的经验和所学,综述非靶向代谢组学的数据分析方法。
本文可结合另一篇博客(代谢组学数据分析的统计学方法综述)一起阅读,以便加深理解。
概述
什么是“代谢组学”(metabolomics)呢?
首先,我们得明确什么叫“代谢物”(metabolite)。维基百科的定义:A metabolite is the intermediate end product of metabolism. The term metabolite is usually restricted to small molecules. 百度百科的定义:代谢物亦称中间代谢物,是指通过代谢过程产生或消耗的物质,生物大分子不包括在内。
目前METLIN数据库中的标准代谢物分子总共超过200,000 种;一般非靶向代谢组学使用质谱仪能检测到人体血液中的代谢信号峰大约接近10,000个。由此可知,代谢组学的特征维度是比较大的。
其次,我们了解下什么叫“代谢组”(metabolome)。维基百科的定义:The metabolome refers to the complete set of small-molecule chemicals found within a biological sample. The biological sample can be a cell, a cellular organelle, an organ, a tissue, a tissue extract, a biofluid or an entire organism. 百度百科的定义:代谢组是指生物体内源性代谢物质的动态整体。而传统的代谢概念既包括生物合成,也包括生物分解,因此理论上代谢物应包括核酸、蛋白质、脂类生物大分子以及其他小分子代谢物质。但为了有别于基因组、转录组和蛋白质组,代谢组目前只涉及相对分子质量约小于1000的小分子代谢物质。
那么“代谢组学”(metabolomics)怎么定义呢?维基百科上说:Metabonomics is defined as "the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification". 百度百科的解释是:代谢组学是效仿基因组学和蛋白质组学的研究思想,对生物体内所有代谢物进行定量分析,并寻找代谢物与生理病理变化的相对关系的研究方式,是系统生物学的组成部分。注意,代谢组学还有个英文写法是“metabonomics”,这两个写法都是可以的,但其实这两个词的侧重点有些区别,此处不深究,感兴趣的童鞋可以自行查找资料了解。
代谢组学从研究特点上可分为非靶向代谢组学和靶向代谢组学。非靶向代谢组学无偏向地检测样本中所有能检测到的代谢物分子,是通过生信方法进行差异分析和通路分析,寻找生物标志物,初步建立模型或代谢物Panel的组学方法。而靶向代谢则是针对特定的代谢物进行检测,由于其使用标准品,因此可以实现代谢物的绝对定量(非靶向代谢组学只能相对定量)。
用于代谢组学研究的样本,主要包括:组织、血液、尿液等,其他如生物体液、分泌物或排泄物也常用于代谢组学研究。
数据采集的方法上来看,主要分为:核磁共振(NMR)、气质联用(GC-MS)及液质联用(LC-MS)。NMR的灵敏度最低,LC-MS的灵敏度最高(可以检测到更多的代谢物)。采集的数据经过处理,可转化成各个代谢信号峰的相对含量值表(常使用XCMS等工具进行处理)。
总的来说,完整的代谢组学研究,应包括实验设计、样本处理、数据采集、数据分析这几个部分,本文仅介绍非靶向代谢组学的数据分析部分(注:本人接触的是血标本的LC-MS数据)。
数据预处理
采集的数据经过处理,可转化成各个信号峰的相对含量值表,这个表一般形式为:每一行代表一个信号(可由RT[保留时间]和m/z[质荷比]确定一个信号峰)在各个样本中的相对含量,也就是说,每一列代表每个样本中各个信号的性对含量(前几列除外,表示各信号的RT、m/z等信息)。每个信号可用RT值和m/z值组合进行命名。
对于得到的这个表,我们常常进行如下3个预处理操作:信号峰注释、标准化校正、质控。
信号峰的注释。可以对同位素峰、加合物峰进行注释,甚至可以初步鉴定部分信号峰所对应的代谢物名称。
标准化校正。可分为批次内校正和批次间校正。需要校正是因为仪器不稳定等情况,可能使信号峰的相对含量出现误差。校正的方法有几种,目前一般首选基于QC样本的标准化方法,即:将所要采集的所有样本取等量混合起来,组成QC样本,然后在采集数据的时候,每隔一定数量的样品,插放一份QC样本。因为QC样本都是一样的,因此可以用QC样本来反映数据采集过程中信号的偏移规律。校正的工具,目前主要推荐中科院ZhuLab开源的MetNormalizer(朱正江研究员的博士生申小涛师兄开发)。
质控。对每个信号峰的QC样本求RSD(相对标准偏差),通常需舍弃RSD超过30%的信号峰(数据质量太差)。
统计分析
单变量分析
二分类问题的单变量分析主要分为:Wilcoxon秩和检验(或 t检验)和 Fold Change分析。多分类问题可能需要ANOVA等方法。常用的可视化方法为 Volcano Plot (火山图),可初步筛选出同时满足Wilcoxon检验统计学差异和Fold Change倍数差异的信号峰。单变量分析很简单,但常常很有效。
值得注意的一点是,单变量统计学检验,其p值的阈值设定,严格来说不应该设定为0.05,需要进行FDR校正(高维数据进行多次假设检验,容易产生大量的假阳性)。但作为初筛,许多研究往往卡得比较松。
单变量分析中,采用中位数还是平均数来代表一个组的值呢?比如计算FC时,是用两组的中位数计算FC还是用均数去计算FC呢,以及统计学检验使用t检验还是选择wilcoxon检验呢?一般来说,如果数据分布是正态分布,则用均数,否则用中位数。
慎用FC值(个人观点):随便使用FC值去筛选变量,很可能导致重要变量被筛出局,举个栗子:
代谢物X在A组15个病例中的峰值分别是:92,95,95,96,96,97,98,100,101,101,101,102,102,103,103,中位数或平均数大致为100;
代谢物X在B组15个病例中的峰值分别是:106,107,108,108,108,108,109,110,111,112,112,112,113,113,115,中位数或平均数大致为110。
代谢物X的FC值(B/A)为1.1。若此时设定FC值以1.2作为界值,X将被排除出模型;然而X可能是一个很好的biomarker,无辜出局。
那么,何时用FC值呢?FC值方法有个特点:FC值越接近1的变量,成为好的biomarker的概率越低。也就是说,噪音变量特别多的时候,采用FC值去排除噪音变量的效率很高。亦即信噪比很低时,FC很管用。所以在特征特别多的任务中,初筛变量的第一步会用FC爽一爽。但若建模效果不理想,有可能是初筛时排除了有效的特征,这个时候应该回过头来放宽界值甚至去除FC标准。
P值是否也需要注意?相对来说,初筛时p值还算靠谱,宽松时可以不进行FDR校正,卡在0.05也还OK。刚刚说的FC值法,实际上触发了假阴性的情况,那么p值其实也有类似情况,当选用非参数检验时,假阴性率会上升。因此慎用非参数检验方法。同样的道理,若初筛后发现建模效果不理想,可以回过头来放宽界值甚至选择统计学检验效能更强的方法。
多元统计分析
多变量分析之前,需要对变量进行标准化(包括中心化和尺度化),尺度化的方法主要有以下两种。
Auto scaling:自动标度化,也叫UV scaling(univariate scaling,单变量标准化),也就是中心化后除以该变量的标准差,也叫Z-score标准化。
Pareto scaling:柏拉图标准化,一般写成Par标准化,与UV scaling的不同之处就是对标准差开根号。
一般用的较多的是Z-score标准化。
多元统计分析非常重要的一步是降维。提到降维,很多人的反应便是PCA、LASSO、PLS等方法。代谢组学中较多使用PLS(偏最小二乘法),因为信号峰之间的相关性较高,LASSO降维不仅会将意义较小的变量剔除,也会将相关性较高(共线性)的变量中剔除多余的。一般代谢组学需要探索代谢物之间的互作与研究结局变量的关系,因此PLS更受欢迎。当然,根据研究目的的不同(比如单纯为了找显著价值的互相独立的biomarker),也可以使用LASSO等方法降维。而PCA作为无监督的方法,在代谢组学中主要仅用于质控或寻找天然的分组。
此处对PLS进行简略介绍(详细介绍可参考博客:偏最小二乘法 Partial Least Squares)。
PLS作为监督学习的一种方法,不仅对自变量x成分进行了映射处理,还对结局变量y进行逐步残差拟合。除了PLS,还有其加强算法——OPLS,区分能力略微更强,可视化效果略微更好。
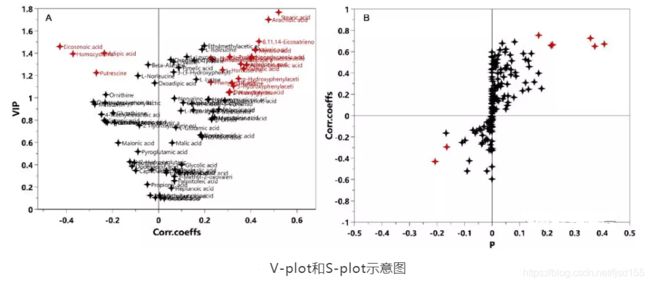
PLS/OPLS的得分图类似于PCA的得分图,但是PLS/OPLS还可对每个变量(特征)求一个VIP值(Variable Importance in Projection),反应的是每个变量对模型解释的贡献度,VIP越大的变量越重要。
除了VIP值,还可以求最终模型中各变量的系数(又称PLS-BETA值)和Corr.Coeffs,以及二者对应的p值。
可综合VIP值和Corr.Coeffs值筛选变量(V-Plot),或者综合PLS-BETA值和Corr.Coeffs值筛选变量(S-Plot)。
评价(O)PLS-DA 模型拟合效果使用R2X、R2Y和Q2Y这三个指标,这些指标越接近1 表示PLS-DA 模型拟合数据效果越好。其中,R2X 和R2Y 分别表示PLSDA分类模型所能够解释X 和Y 矩阵信息的百分比,Q2Y 则为通过交叉验证计算得出,用以评价PLS-DA模型的预测能力,Q2Y 越大代表模型预测效果较好。
PCA分析中R2X >0.4为好;PLS-DA 和 OPLS-DA分析中,R2X 这个参数不重要了,主要是R2Y 和Q2,这两个值>0.5 为好,越接近1越好。OPLS-DA中Q2(cum),是指建模后模型的预测能力,以大于0.5为宜,越接近1越好,cum 表示累积的意思。
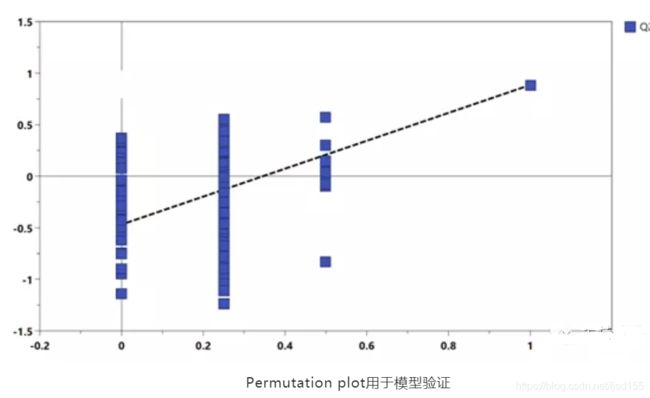
对于PLS/OPLS,我们常常需进行 permutation test(置换检验)(勿与交叉检验混淆),以确定模型是否过拟合。一般需检验模型的Q2值和R2值(Q2)。对于Q2,要求置换检验结果的在y轴上的截距小于0,方可认为模型没有过拟合。置换检验的基本原理:将真实分类结果(标签)屏蔽,重新随机赋予分类结果(标签),再进行建模。如果真实建模的Q2和随机标签建模的Q2接近,则说明模型过拟合。具体原理请参考其他资料。置换检验可视化的图,横坐标表示的是置换后的标签与真实标签的相关性(有多少比例的样本未打乱重新赋予标签)。
进行降维后,除了使用PLS/OPLS多元分析方法可以继续进行多元统计建模外,还可使用SVM、RandomFores、ANN等方法进行建模。另外,最终最好使用Logistic回归建立具备临床(或生物学)解释意义的模型。
另外,瑞典查尔默斯理工大学的施琳大神前不久发表在bioinformatics上的一篇文章,介绍了一个用于多元统计分析的方法,并开发了一个R包MUVR。
物质鉴定
对于质谱仪测定的代谢物,有公共数据库可以根据m/z等信息进行鉴定,如HMDB,MassBank,METLIN等。
有时候需要先对两批数据中取交集,这个时候可以根据m/z值和RT值进行确定,比如同时满足容差条件:m/z在5ppm内,RT在50内。之后还可根据二级谱图(MS-MS)的信息,进一步确定。
关于ppm,举个栗子(摘自:代谢组学研究中需要了解的质谱知识丨质量精度):
C6H12O6理论精确分子量为180.0634
如果测得分子量为180.0631,则误差为
180.0631-180.0634=-0.0003Da=-0.3mDa
(180.0631-180.0634)/180.0634=1.67e-6 即 1.67ppm
网络分析
包括富集分析(Enrichment analysis)和通路分析(Pathway analysis)。通路分析中添加了通路的拓扑分析,输出通路在整体网络中的重要性(impact),重要性越大,可能意味着在整个通路中的地位越核心,那么从impact值也可以反映出来。
致谢
感谢申小涛大神、施琳大神和陈显扬大神等前辈曾给予指点!
参考资料
非靶向代谢组学数据分析总结-纲要
History of Metabolomics
维基百科相应词条
百度百科相应词条
麦特绘谱-代谢组学数据处理
代谢组学精华汇总及该博文的参考资料