Bioinformatics

Abstract
The Gene Network Estimation Tool (GNET) is designed to build gene regulatory networks from transcriptomic gene expression data with a probabilistic graphical model. The data preprocessing, model construction, and visualization modules of the original GNET software were developed on different programming platforms, which were inconvenient for users to deploy and use. Here we present GNET2, an improved implementation of GNET as an integrated R package. GNET2 provides more flexibility for parameter initialization and regulatory module construction based on the core iterative modeling process of the original algorithm. The data exchange interface of GNET2 is handled within an R session automatically. Given the growing demand for regulatory network reconstruction from transcriptomic data, GNET2 offers a convenient option for gene regulatory network inference on large datasets
Brief:转录因子及其调控基因推断,R package

Abstract
Cancer Gene and Pathway Explorer (CGPE) is developed to guide biological and clinical researchers, especially those with limited informatics and programming skills, performing preliminary cancer related biomedical research using transcriptional data and publications. CGPE enables three user-friendly online analytical and visualization modules without requiring any local deployment. The GenePub HotIndex applies natural language processing, statistics, and association discovery to provide analytical results on gene-specific PubMed publications, including gene-specific research trends, cancer types correlations, top-related genes, and the WordCloud of publication profiles. The OnlineGSEA enables Gene Set Enrichment Analysis (GSEA) and results visualizations through an easy-to-follow interface for public or in-house transcriptional datasets, integrating the GSEA algorithm and preprocessed public TCGA and GEO datasets. The preprocessed datasets ensure gene sets analysis with appropriate pathway alternation and gene signatures. The CellLine Search presents evidence-based guidance for cell line selections with combined information on cell line dependency, gene expressions, and pathway activity maps, which are valuable knowledge to have before conducting gene-related experiments. In a nutshell, the CGPE webserver provides a user-friendly, visual, intuitive, and informative bioinformatics tool that allows biomedical researchers to perform efficient analyses and preliminary studies on in-house and publicly available bioinformatics data.
Brief:癌症细胞系数据库,共三个功能“:1根据基因名提供该基因在各癌症的研究并提供pubmed链接;2集成公共数据,根据提供基因集进行在线GSEA;3根据基因名检索个癌细胞系对该基因依赖性及基因表达量

Abstract
The cost of drug development has dramatically increased in the last decades, with the number new drugs approved per billion US dollars spent on R&D halving every year or less. The selection and prioritization of targets is one the the most influential decisions in drug discovery. Here we present a Gaussian Process model for the prioritization of drug targets cast as a problem of learning with only positive and unlabeled examples.
Brief:提出一种基于单类高斯过程( One Class Gaussian Processes)的机器学习方法通过与已批准药物标靶的相似性对候选蛋白进行排序,同时提出一个超参数的选择方法,基本内涵是为每个训练样本使用不同超参数,给予密集区域的样本更多的权重(这个超参数主要解决了此类学习问题只有正例样本因此难以根据边际似然最大化来选择超参数的问题)__仅正例机器学习模型训练

Abstract
One major goal of single-cell RNA sequencing (scRNAseq) experiments is to identify novel cell types. With increasingly large scRNAseq datasets, unsupervised clustering methods can now produce detailed catalogues of transcriptionally distinct groups of cells in a sample. However, the interpretation of these clusters is challenging for both technical and biological reasons. Popular clustering algorithms are sensitive to parameter choices, and can produce different clustering solutions with even small changes in the number of principal components used, the k nearest neighbor, and the resolution parameters, among others.
Brief:多次对单细胞各样本进行向下采样,并重新聚类后计算各聚类间的 Jaccard index 以此评估单细胞聚类的稳定性


Abstract
Gene expression and regulation, a key molecular mechanism driving human disease development, remains elusive, especially at early stages. Integrating the increasing amount of population-level genomic data and understanding gene regulatory mechanisms in disease development are still challenging. Machine learning has emerged to solve this, but many machine learning methods were typically limited to building an accurate prediction model as a “black box”, barely providing biological and clinical interpretability from the box.
To address these challenges, we developed an interpretable and scalable machine learning model, ECMarker, to predict gene expression biomarkers for disease phenotypes and simultaneously reveal underlying regulatory mechanisms. Particularly, ECMarker is built on the integration of semi- and discriminative- restricted Boltzmann machines, a neural network model for classification allowing lateral connections at the input gene layer. This interpretable model is scalable without needing any prior feature selection and enables directly modeling and prioritizing genes and revealing potential gene networks (from lateral connections) for the phenotypes. With application to the gene expression data of non-small cell lung cancer (NSCLC) patients, we found that ECMarker not only achieved a relatively high accuracy for predicting cancer stages but also identified the biomarker genes and gene networks implying the regulatory mechanisms in the lung cancer development. Additionally, ECMarker demonstrates clinical interpretability as its prioritized biomarker genes can predict survival rates of early lung cancer patients (p-value < 0.005). Finally, we identified a number of drugs currently in clinical use for late stages or other cancers with effects on these early lung cancer biomarkers, suggesting potential novel candidates on early cancer medicine.
**Brief:基于半受限玻尔兹曼机( semi-restricted Boltzmann machines)构建早期癌症判别模型,但相对于其他机器学习模型仅有高预测精度但无临床解释性特点,ECMarker提供基因的判别权重,因此具有可解释性
**

Abstract
The advent of high-throughput technologies has provided researchers with measurements of thousands of molecular entities and enable the investigation of the internal regulatory apparatus of the cell. However, network inference from high-throughput data is far from being a solved problem. While a plethora of different inference methods have been proposed, they often lead to non-overlapping predictions, and many of them lack user-friendly implementations to enable their broad utilisation. Here we present COSIFER, a package and a companion web-based platform to infer molecular networks from expression data using state-of-the-art consensus approaches. COSIFER includes a selection of state-of-the-art methodologies for network inference and different consensus strategies to integrate the predictions of individual methods and generate robust networks.
Brief:集成10中不同的网络推断方法,提供web及python包;允许使用一个或多个方法集成推断,集成推断通过现有三种框架整合1)WOC;2) WOC (hard);3)Similarity Network Fusion (SNF)

Abstract
Breast cancer is a highly heterogeneous disease, and there are many forms of categorization for breast cancer based on gene expression profiles. Gene expression profiles are variables and may show differences if measured at different time points or under different conditions. In contrast, biological networks are relatively stable over time and under different conditions. In this study, we used a gene interaction network from a new point of view to explore the subtypes of breast cancer based on individual-specific edge perturbations measured by relative gene expression value. Our study reveals that there are four breast cancer subtypes based on gene interaction perturbations at the individual level. The new network-based subtypes of breast cancer show strong heterogeneity in prognosis, somatic mutations, phenotypic changes and enriched pathways. The network-based subtypes are closely related to the PAM50 subtypes and immunohistochemistry index. This work helps us to better understand the heterogeneity and mechanisms of breast cancer from a network perspective.
Brief:提供了一种edge-based的生物信息学分析方法:将基因表达矩阵转换成Rank形式后,通过计算背景网络的edge的rank以代表基因互作的扰动情况,最后将癌症样本的rank矩阵减去正常样本的扰动矩阵从而归一化;implemented in R and is available at https://github.com/Marscolono/SSPGI.git


Abstract
The tumor immune microenvironment is a main contributor to cancer progression and a promising therapeutic target for oncology. However, immune microenvironments vary profoundly between patients and biomarkers for prognosis and treatment response lack precision. A comprehensive compendium of
tumor immune cells is required to pinpoint predictive cellular states and their spatial localization. We generated a single-cell tumor immune atlas, jointly analyzing >500,000 cells from 217 patients and 13 cancer types, providing the basis for a patient stratification based on immune cell compositions. Projecting
immune cells from external tumors onto the atlas facilitated an automated cell annotation system for a harmonized interpretation. To enable in situ mapping of immune populations for digital pathology, we applied SPOTlight, combining single-cell and spatial transcriptomics data and identifying striking spatial
immune cell patterns in tumor sections. We expect the tumor immune cell atlas, together with our versatile toolbox for precision oncology, to advance currently applied stratification approaches for prognosis and immuno-therapy.
Brief:整合了50w泛癌免疫细胞数据,并可作为query数据集用于annotation


Abstract
Perturbation biology is a powerful approach to modeling quantitative cellular behaviors and understanding
detailed disease mechanisms. However, large-scale protein response resources of cancer cell lines to per�turbations are not available, resulting in a critical knowledge gap. Here we generated and compiled perturbed expression profiles of �210 clinically relevant proteins in >12,000 cancer cell line samples in response to 170 drug compounds using reverse-phase protein arrays. We show that integrating perturbed protein response signals provides mechanistic insights into drug resistance, increases the predictive power for drug sensitivity, and helps identify effective drug combinations. We build a systematic map of ‘‘protein�drug’’ connectivity and develop a user-friendly data portal for community use. Our study provides a rich resource to investigate the behaviors of cancer cells and the dependencies of treatment responses, thereby enabling a broad range of biomedical applications.
Brief:大规模药物-癌症细胞系扰动反向蛋白微阵列数据



Brief:通过图卷积网络反卷积空间转录组数据已会服空间范畴的表型


Genome Research
Abstract
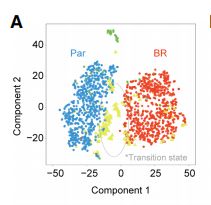
Single-cell RNA-seq’s (scRNA-seq) unprecedented cellular resolution at a genome-wide scale enables us to address questions about cellular heterogeneity that are inaccessible using methods that average over bulk tissue extracts. However, scRNA-seq data sets also present additional challenges such as high transcript dropout rates, stochastic transcription events, and complex population substructures. Here, we present a single-cell RNA-seq analysis and klustering evaluation (SAKE), a robust method for scRNA-seq analysis that provides quantitative statistical metrics at each step of the analysis pipeline. Comparing SAKE to multiple single-cell analysis methods shows that most methods perform similarly across a wide range of cellular contexts, with SAKE outperforming these methods in the case of large complex populations. We next applied the SAKE algorithms to identify drug-resistant cellular populations as human melanoma cells respond to targeted BRAF inhibitors (BRAFi). Single-cell RNA-seq data from both the Fluidigm C1 and 10x Genomics platforms were analyzed with SAKE to dissect this problem at multiple scales. Data from both platforms indicate that BRAF inhibitor-resistant cells can emerge from rare populations already present before drug application, with SAKE identifying both novel and known markers of resis�tance. These experimentally validated markers of BRAFi resistance share overlap with previous analyses in different mela�noma cell lines, demonstrating the generality of these findings and highlighting the utility of single-cell analysis to elucidate mechanisms of BRAFi resistance.
Brief:通过非负矩阵因子分解对单细胞数据进行聚类后分析并搭建了分析流程,SAKE;将其运用到癌细胞的耐药研究发现了潜在的耐药细胞群体,提示非负因子矩阵分解在解析细胞异质性的能力上一定程度优于直接基于基因表达量的聚类方法

Abstract
Single-cell RNA sequencing (scRNA-seq) is a versatile tool for discovering and annotating cell types and states, but the determination and annotation of cell subtypes is often subjective and arbitrary. Often, it is not even clear whether a given cluster is uniform. Here we present an entropy-based statistic, ROGUE, to accurately quantify the purity of identified cell clusters. We demonstrate that our ROGUE metric is broadly applicable, and enables accurate, sen�sitive and robust assessment of cluster purity on a wide range of simulated and real datasets. Applying this metric to fibroblast, B cell and brain data, we identify additional subtypes and demonstrate the application of ROGUE-guided analyses to detect precise signals in specific subpopulations. ROGUE can be applied to all tested scRNA-seq datasets, and has important
implications for evaluating the quality of putative clusters, discovering pure cell subtypes and constructing comprehensive, detailed and standardized single cell atlas
Brief:提供一个熵依赖的度量以衡量单细胞亚群的“纯度”,不同平台,不同测序深度均不会影响其衡量准确度


Abstract
Identifying gene expression programs underlying both cell-type identity and cellularactivities (e.g. life-cycle processes, responses to environmental cues) is crucial for understanding the organization of cells and tissues. Although single-cell RNA-Seq (scRNA-Seq) can quantify transcripts in individual cells, each cell’s expression profile may be a mixture of both types of programs, making them difficult to disentangle. Here, we benchmark and enhance the use of matrix factorization to solve this problem. We show with simulations that a method we call consensus non-negative matrix factorization (cNMF) accurately infers identity and activity programs, including their relative contributions in each cell. To illustrate the insights this approach enables, we apply it to published brain organoid and visual cortex scRNA-Seq datasets; cNMF refines cell types and identifies both expected (e.g. cell cycle and hypoxia) and novel activity programs, including programs that may underlie a neurosecretory phenotype and synaptogenesis. DOI: https://doi.org/10.7554/eLife.43803.001
Brief:提出一种优化的非负矩阵分解的方法,主要思想是多次进行矩阵因子分解并对结果进行平均从而提高稳健性,运用于单细胞数据解析非细胞亚群特异性的细胞激活程序表达


Abstract
Efficient single-cell assignment without prior marker gene annotations is essential for single-cell sequencing data analysis. Current methods, however, have limited effectiveness for distinct single-cell assignment. They failed to achieve a well-generalized performance in different tasks because of the inherent heterogeneity of different single-cell sequencing datasets and different single-cell types. Furthermore, current methods are inefficient to identify novel cell types that are absent in the reference datasets. To this end, we present scLearn, a learning-based framework that automatically infers quantitative measurement/similarity and threshold that can be used for different single-cell assignment tasks, achieving a well-generalized assignment performance on different single-cell types. We evaluated scLearn on a comprehensive set of publicly available benchmark datasets. We proved that scLearn outperformed the comparable existing methods for single-cell assignment from various aspects, demonstrating state-of-the-art effectiveness with a reliable and generalized single-cell type identification and categorizing ability
Brief:基于学习的reference依赖的单细胞注释框架,除了ref的识别准确外,在识别unsigned cell type的情况表现良好,并且可以基于多重标签,进行校正后分配出细胞类型外标签的能力

Brief:解释COVID-19中脊索萎缩相关基因SMN1可能对新冠肺炎诱导的肺损伤有保护作用


Abstract
The functional specialization of cell types arises during development and is shaped by cell–cell communication networks determining a distribution of functional cell states that are collectively important for tissue functioning. However, the identification of these tissue-specific functional cell states remains challenging. Although a plethora of computational approaches have been successful in detecting cell types and subtypes, they fail in resolving tissue-specific functional cell states. To address this issue, we present FunRes, a computational method designed for the identification of functional cell
states. FunRes relies on scRNA-seq data of a tissue to initially reconstruct the functional cell–cell communication network, which is leveraged for partitioning each cell type into functional cell states. We applied FunRes to 177 cell types in 10 different tissues and demonstrated that the detected states correspond to known functional cell states of various cell types, which cannot be recapitulated by existing computational tools. Finally, we characterize emerging and vanishing functional cell states in aging and disease, and demonstrate their involvement in key tissue functions. Thus, we believe that FunRes will be of great utility in the characterization of the functional landscape of cell types and the identification of dysfunctional cell states in aging and disease.
Brief:通过细胞通讯及转录因子等信息重构矩阵分析细胞状态


Abstract
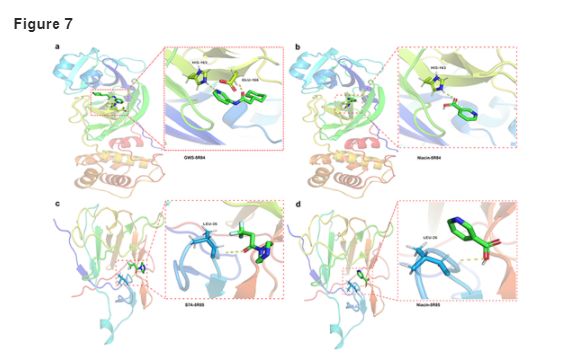
We revealed the clinical characteristics of CRC patients and COVID-19 patients, including predisposing genes, survival rate and prognosis. Moreover, the results of molecular docking analysis indicated that niacin exerted effective binding capacity in COVID-19. Further, we disclosed the targets, biological functions and signaling pathways of niacin in CRC/COVID-19. The analysis indicated that niacin could help in treating CRC/COVID-19 through cytoprotection, enhancement of immunologic functions, inhibition of inflammatory reactions and regulation of cellular microenvironment. Furthermore, five core pharmacological targets of niacin in CRC/COVID-19 were also identified, including BCL2L1, PTGS2, IL1B, IFNG and SERPINE1.
Brief:结直肠癌COVID-19感染风险增加,且由于免疫能力低下及缺乏抗COVID-19药物,需要为其筛选可选药物:1鉴定COVID相关基因在CRC患者的差异表达;2差异基因基于多变量COX回归定义风险分数;3数据库搜索烟酸药理靶点后发现与COVID-19_CRC基因集有14个overlap;4String确定5个核心基因;5分子对接,提示烟酸和核心基因之一IL1B亲和力较高

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is accountable for the cause of coronavirus disease (COVID-19) that causes a major threat to humanity. As the spread of the virus is probably getting out of control on every day, the epidemic is now crossing the most dreadful phase. Idiopathic pulmonary fibrosis (IPF) is a risk factor for COVID-19 as patients with long-term lung injuries are more likely to suffer in the severity of the infection. Transcriptomic analyses of SARS-CoV-2 infection and IPF patients in lung epithelium cell datasets were selected to identify the synergistic effect of SARS-CoV-2 to IPF patients. Common genes were identified to find shared pathways and drug targets for IPF patients with COVID-19 infections. Using several enterprising Bioinformatics tools, protein–protein interactions (PPIs) network was designed. Hub genes and essential modules were detected based on the PPIs network. TF-genes and miRNA interaction with common differentially expressed genes and the activity of TFs are also identified. Functional analysis was performed using gene ontology terms and Kyoto Encyclopedia of Genes and Genomes pathway and found some shared associations that may cause the increased mortality of IPF patients for the SARS-CoV-2 infections. Drug molecules for the IPF were also suggested for the SARS-CoV-2 infections.
Brief:肺纤维化是COVID-19发病危险因素,因此探索肺纤维化及COVID-19患者转录组,鉴定了相应miRNA-TF调控网络并鉴定网络中hub基因,在数据库中鉴定了可能的治疗药物(DSigDB)