参考:碱基矿工
从零开始完整学习全基因组测序数据分析:第4节 构建WGS主流程
GATK4.0和全基因组数据分析实践(上)
前言
由于人类基因组数据过大(我的小小服务器也只有50G),而人的参考序列长度就有3Gb,且高深度测序结果往往动辄100G。因此作者也尝试用E.coli K12 的数据作为替代,一来它的基因不是很复杂,数据小,它的基因组长度只有4.6Mb,二来也能很好的跑一遍流程,熟悉过程。
咱们开始吧~
准备阶段
首先需要在服务器上部署相关的软件,包括以下四个软件。
1)BWA (Burrow-Wheeler Aligner):非常权威的NGS数据比对软件,https://github.com/lh3/bwa。
2)Samtools:一个专门用于处理比对数据的软件,https://github.com/samtools/samtools。
3)Picard:也是一款功能强大的NGS数据处理工具,功能和samtools 很类似,但更多的是与其互补,http://broadinstitute.github.io/picard/。
4)GATK:是一款非常权威的基因数据变异检测工具。目前有3.x和4.x两个不同的版本。4.x在核心算法层面并没太多的修改,采用了新的设计模式,后续GATK团队也将主要维护4.x的版本。但目前还不如3.x 版本稳定,且短期来看3.x版本还将在业内继续使用一段时间。这里作者也以GATK3.8(最新版本)作为流程的重要工具进行分析流程的构建。https://software.broadinstitute.org/gatk/download/
对于WGS 分析来说,这四个工具就够了。
安装
关于conda 安装,参见:https://www.jianshu.com/p/632f35c48137
BWA 的安装
conda create -n wgs python=2 bwa # 创建环境及conda 安装
source activate wgs # 激活环境
# 这样我们安装的包就只在这一个环境下了,不会使linux 工作环境污染。
samtools
conda install samtools
Picard
conda install picard
GATK
conda install gatk
sratoolkit
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.10.5/sratoolkit.2.10.5-centos_linux64.tar.gz # 下载
添加内容的环境变量,vi ~/.bashrc 并在底部加上文件目录
db-config --interactive # 按照提示完成配置
项目结构设计
./20200524_wgs_practice/
├── bin
├── input
└── output
- bin 存放所有执行程序和代码
- input 存放所有输入数据
- output 存放所有输出数据
获取E.coli K12的参考基因组序列
一般来说基因组信息都可以从ensemble 或者ncbi两个数据库获取,除此之外也还有很多的数据库,可以参考以下:
https://www.jianshu.com/p/6c45620b2578
-
这里我是通过ncbi获取。
可以通过ftp下载得到fasta 格式的序列。
这里我们可以利用wget 获得fasta 文件
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/005/845/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna.gz
通过gzip解压缩并将名字重命名为E.coli_K12_MG1655.fa。
gzip -dc GCF_000005845.2_ASM584v2_genomic.fna.gz > E.coli_K12_MG1655.fa
我们可以查看一下它的前十行
$head -10 E.coli_K12_MG1655.fa
>NC_000913.3 Escherichia coli str. K-12 substr. MG1655, complete genome
AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGCTTCTGAACTG
GTTACCTGCCGTGAGTAAATTAAAATTTTATTGACTTAGGTCACTAAATACTTTAACCAATATAGGCATAGCGCACAGAC
AGATAAAAATTACAGAGTACACAACATCCATGAAACGCATTAGCACCACCATTACCACCACCATCACCATTACCACAGGT
AACGGTGCGGGCTGACGCGTACAGGAAACACAGAAAAAAGCCCGCACCTGACAGTGCGGGCTTTTTTTTTCGACCAAAGG
TAACGAGGTAACAACCATGCGAGTGTTGAAGTTCGGCGGTACATCAGTGGCAAATGCAGAACGTTTTCTGCGTGTTGCCG
ATATTCTGGAAAGCAATGCCAGGCAGGGGCAGGTGGCCACCGTCCTCTCTGCCCCCGCCAAAATCACCAACCACCTGGTG
GCGATGATTGAAAAAACCATTAGCGGCCAGGATGCTTTACCCAATATCAGCGATGCCGAACGTATTTTTGCCGAACTTTT
GACGGGACTCGCCGCCGCCCAGCCGGGGTTCCCGCTGGCGCAATTGAAAACTTTCGTCGATCAGGAATTTGCCCAAATAA
AACATGTCCTGCATGGCATTAGTTTGTTGGGGCAGTGCCCGGATAGCATCAACGCTGCGCTGATTTGCCGTGGCGAGAAA
下载测序数据
ncbi 的SRA ,Sequence Read Archive,数据库http://www.ncbi.nlm.nih.gov/sra/ 是目前最常用的存储测序数据的数据库。
E.coli K12 作为一种非常常用的模式生物,已经有很多测序信息在ncbi上了,这里选择其中一个测序信息SRR1770413。
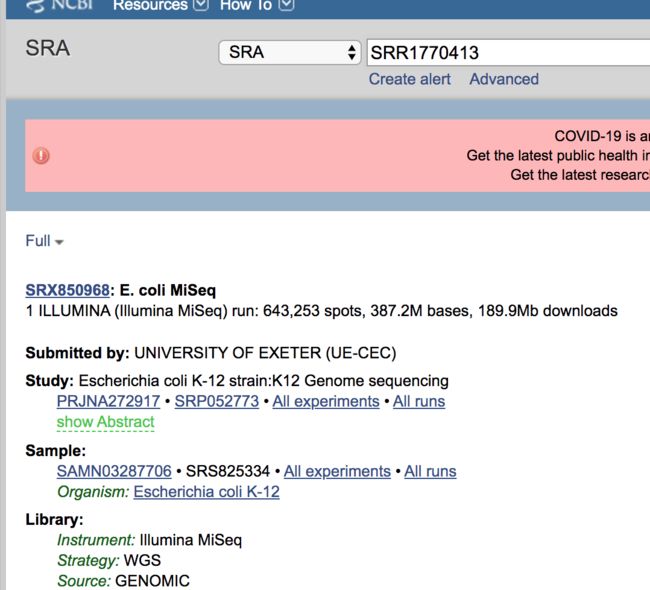
这个数据来自Illumina MiSeq测序平台,read长度为300bp,测序类型是Pair-end。
小插曲,PE测序与SE测序
在之前的测序技术的介绍中,我们已经知道包括illumina 在内的NGS测序都是短读长的测序,每次测出来的read 长度都不太长,为了尽可能的把DNA的序列片段多测出来,一边测不够,就测两边。
于是就有了一种从被测DNA 序列两端各测序一次的模式,被称为双末端测序(Pair-End Sequencing,简称PE测序)。
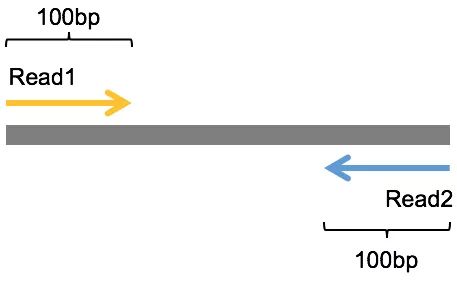
灰色的是被测序的DNA 片段,左边黄色与右边蓝色分别是测序出来的read1 与read2 序列,上图中假定它们的长度都是100bp。
虽然很多时候Pair-End测序还是无法完全的将这个被测DNA 的片段完全测完,但它依然提供了非常有用的信息。
比如当我们知道每一对的read1与read2 都来自于同一个DNA片段,read1 与read2 之间的距离是这个DNA片段的长度,且read1 与read2 的方向是刚好相反的。(这些信息对于后期的变异检测等分析非常有用)
除此之外,read1 在fq1 文件中位置和read2 在fq2 文件中位置是相同的,而且read ID 之间只在末尾有一个'/1'或者'/2'的差别。
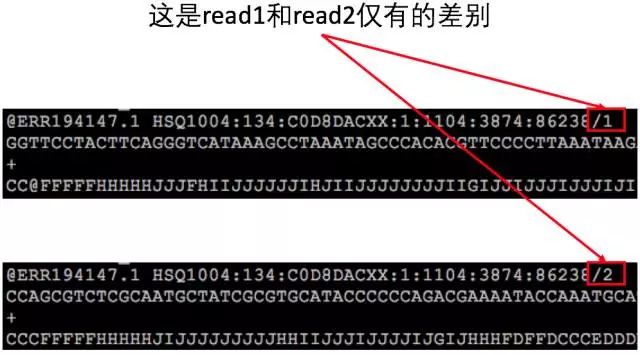
对应双末端,自然也有单末端测序(Single End Sequecing,简称SE测序),它只测序其中一端。因此,在使用如bwa进行比对时,in2.fq是非强制性的(所以用方括号括起来),只有在双末端测序时才需要添加。
Usage: bwa mem [options] [in2.fq]
继续下载SRA 文件
我们可以在通过sratoolkit 工具直接下载prefetch SRRxxxxx某个SRR 的run 编号。(但实测速度非常慢)
或者也可以在NCBI 的SRR 网站上下载相关的数据。
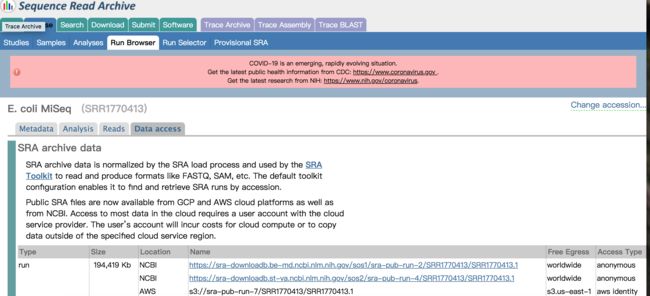
下载完成后由于SRR 得到的是压缩的文件,我们需要把里面的read1和read2的测序数据解压出来,变成需要的fastq格式。
fastq-dump --split-files SRR1770413.1
准备就绪,现在目录下就有这些东西了~
$tree ..
..
|-- bin
|-- input
| |-- E.coli_K12_MG1655.fa
| |-- E.coli_K12_MG1655.fa.fai
| |-- GCF_000005845.2_ASM584v2_genomic.fna.gz
| |-- SRR1770413.1
| |-- SRR1770413.1_1.fastq
| `-- SRR1770413.1_2.fastq
`-- output
正式开始
-
将目录内容打理一下,再来看看有哪些东西。

质控
关于质控,是不可以省略的。
可以借助Trimmomatic。
参见https://www.jianshu.com/p/56977fc52ecf
构建索引
首先我们需要为该序列构建一个索引,以便能够在序列比对的时候进行快速的搜索和定位。这里使用samtools。
samtools faidx E.coli_K12_MG1655.fa
根据samtools 帮助界面可知,faidx 还可以对序列内容进行提取。
faidx index/extract FASTA
因而我们也可以用它来提取特定区域的序列内容。
$samtools faidx E.coli_K12_MG1655.fa NC_000913.3:1000000-1000200
>NC_000913.3:1000000-1000200
GTGTCAGCTTTCGTGGTGTGCAGCTGGCGTCAGATGACAACATGCTGCCAGACAGCCTGA
AAGGGTTTGCGCCTGTGGTGCGTGGTATCGCCAAAAGCAATGCCCAGATAACGATTAAGC
AAAATGGTTACACCATTTACCAAACTTATGTATCGCCTGGTGCTTTTGAAATTAGTGATC
TCTATTCCACGTCGTCGAGCG
序列比对
通过NGS 仪器测序得到的,而NGS 在测定这些序列的过程中,非常粗暴地将序列给冲散成一个个短序列(read),保存在FASTQ 文件中。因此文件中不同的read 之间是没有位置关系的。
因此首先要做的就是通过序列比对的方式,将这些read 和物种的参考基因组(比如人的全基因组)比较,从而找到不同read 在参考基因组上本来的位置,将它们捋顺排好。
在这次实践中,我们就需要将获得的E.coli 测序信息,与原始参考基因组序列进行比对,确定每一个read的位置。
即便是相比起双序列比对已经有所优化的BLAST 算法,面对海量的短数据时,执行的耗费的时间也太长了。而这里使用的便是BWA,其将BW(Burrows-Wheeler)压缩算法和后缀树相结合,可以耗费较小的时间与空间,获得较为准确的序列比对结果。
关于blast 的知识:BLAST 序列比对
使用bwa 构建索引
首先需要为参考序列构建bwa 比对所需要的FM-index 比对索引。
$bwa index E.coli_K12_MG1655.fa
[bwa_index] Pack FASTA... 0.03 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 2.27 seconds elapse.
[bwa_index] Update BWT... 0.04 sec
[bwa_index] Pack forward-only FASTA... 0.02 sec
[bwa_index] Construct SA from BWT and Occ... 1.26 sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa index E.coli_K12_MG1655.fa
[main] Real time: 3.904 sec; CPU: 3.625 sec
由于该序列比较短,所以很快(4s 不到,相比来说人类基因组就得要3h了)索引就创建完毕了。
这时文件中便会多出来几个**.fa.** 的文件。ps:其中E.coli_K12_MG1655.fa.fai 是我们之前用samtools 工具获得的。
$ls -l
total 12544
-rw-r--r-- 1 root root 4699745 May 24 10:48 E.coli_K12_MG1655.fa
-rw-r--r-- 1 root root 12 May 27 16:17 E.coli_K12_MG1655.fa.amb
-rw-r--r-- 1 root root 98 May 27 16:17 E.coli_K12_MG1655.fa.ann
-rw-r--r-- 1 root root 4641732 May 27 16:17 E.coli_K12_MG1655.fa.bwt
-rw-r--r-- 1 root root 29 May 24 10:54 E.coli_K12_MG1655.fa.fai
-rw-r--r-- 1 root root 1160415 May 27 16:17 E.coli_K12_MG1655.fa.pac
-rw-r--r-- 1 root root 2320880 May 27 16:17 E.coli_K12_MG1655.fa.sa
接着我们使用mem BWA-MEM algorithm 这个算法,将E.coli K12质控后的测序数据定位到其参考基因组上。ps:一般MEM模块 适合长read的比对,目的是解决第三代测序技术这种能够产生长达几十kb甚至几Mbp的read情况。而通常小于等于70bp 的情况下,一般使用ALN模块。
为了方便显示,就不使用代码块而转为引用。
$bwa mem -t 4 -R '@RG\tID:foo\tPL:illumina\tSM:E.coli_K12' ~/20200524_wgs_practice/input/E.col/fasta/E.coli_K12_MG1655.fa ~/20200524_wgs_practice/input/E.col/fastq/SRR1770413.1_1.fastq ~/20200524_wgs_practice/input/E.col/fastq/SRR1770413.1_2.fastq|samtools view -Sb - > ~/20200524_wgs_practice/output/E_coli_K12.bam && echo "** bwa mapping done **"
解读bwa 语法
由bwa 说明界面可知:
Usage: bwa mem [options] [in2.fq]
[options] 为一系列可选择项,比如这里的-t 4, 表示四线程,-R 则指的是Read Group的字符串信息,以。而
- 特别强调的
-R
@RG\tID:foo\tPL:illumina\[tSM:E.coli_K12'](tsm:E.coli_K12')在比对语法中,我们以@RG开头,表示为对比对的read 进行分组。关于read group有以下几个参数:
1)ID。一般为测序的lane ID。
2)PL,为测序的平台,需要准确填写。在GATK中,PL只被允许写为ILLUMINA,SLX,SOLEXA,SOLID,454,LS454,COMPLETE,PACBIO,IONTORRENT,CAPILLARY,HELICOS或UNKNOWN等信息之一。如果这一步没有设定正确的PL名称,则后续使用GATK过程则可能会出现报错。
3)SM:为样本ID,因为我们在测序时,由于样本量较多,可能会分成许多不同的lane 被分别测出来,这时候可以使用SM 区分这些样本。
4)LB:测序文库的名字。一般如果ID 足够用于区分,则可以不设定LB。
对于序列比对而言,设定这四个参数就足够了。除此之外,在RG中还需要用制表符\t将各个内容区分开。
而[in2.fq] 则表示质控后的fastq 文件(也就是参考序列),因为这是双末端测序,因此其结果有两个文件。
管道后的内容
而其中管道|后面的内容,则是将对比数据引至samtools工具,通过samtools 将文件转换为bam 格式(sam格式的二进制形式),其中samtools view -b便表示输出为bam。另外-Sb与 >中间的-表示被引流的数据。接着重定向> 输出到文件并保存。
对比对结果进行排序
samtools sort -@ 4 -m 4G -O bam -o E_coli_K12.bam E_coli_K12.sorted.bam
其中-@ 4表示排序时的线程,设定为4。
-m表示限制排序时最大的内存消耗,设定为4G。
-O bam表示指定输出格式为bam。
-o表示输出文件名,为‘E_coli_K12.sorted.bam’。
由于这里使用后我的会提示,Error: samtools sort: couldn't allocate memory for bam_mem,尝试不设置内存。
samtools sort -O bam -o E_coli_K12.sorted.bam E_coli_K12.bam
$ll
total 481124
-rw-r--r-- 1 root root 284944979 May 27 16:41 E_coli_K12.bam
-rw-r--r-- 1 root root 207714622 May 27 18:26 E_coli_K12.sorted.bam
去除重复序列
完成了比对之后,就可以开始进行去重复了。(去除PCR 重复序列)
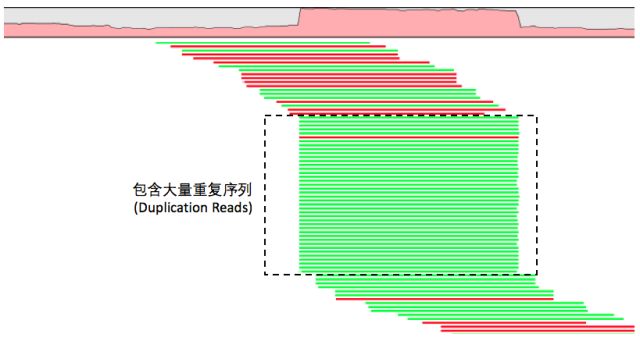
重复序列的产生
首先,什么是重复序列?
我们知道,在进行NGS 测序时,其中一个步骤便是桥式PCR扩增。而进行该步骤目的就是为了弥补测序文库由于构建过程的种种问题而导致浓度不够的缺陷,因而会使测序过程极有可能漏掉某些原本基因组上的一些DNA片段,导致测序不全。
而正因为整个反应都是同步进行,无差别式扩增, 所以某些本身密度就不是很低的DNA序列也会被同步的放大,而这些DNA 片段就非常有可能会被重复去进行测序。
重复序列的主要影响
其主要影响在于增大了变异的结果。
1)DNA 在打断过程可能会引发一些碱基的颠换(嘌呤与嘧啶互换),而PCR 过程会放大该结果,以产生变异。
2)PCR 这一过程本身可能也会带来新的碱基错误。发生在前几轮的PCR 扩增反应发生的错误会在后续的过程里继续放大,带来新的变异。
3)对于真实的变异,PCR 反应可能会对包含某一个碱基的DNA模版扩增而更加剧烈(PCR bias),因此如果反应对参考等位基因的模版偏向的话,可能会导致变异碱基的信号变小,因而导致假阴性结果。
因此,重复序列对于真实的变异检测与真实序列的检测都有不好的影响。GATK、Samtools 这些工具都会默认序列数据为非重复序列,因此重复序列的去除是必不可少的操作。(或者直接采用PCR-free 的方案)
进行重复序列的去除工作
现有的工具包括Samtools、Picard、gatk 都可以用于去除重复序列。
samtools 的rmdup 是直接将重复序列从比对BAM文件中删除。picard与gatk 的MarkDuplicates则是在BAM的FLAG信息中标记出来,不作删除,但我们可以在变异检测时识别它们,并忽略。
- 我们可以用gatk 来创建
gatk MarkDuplicates \
-I E_coli_K12.sorted.bam\
-O E_coli_K12.sorted.markdup.bam \
-M E_coli_K12.sorted.markdup_metrics.txt
$tree .
.
|-- E_coli_K12.bam
|-- E_coli_K12.sorted.bam
|-- E_coli_K12.sorted.markdup.bam
`-- E_coli_K12.sorted.markdup_metrics.txt
0 directories, 4 files
- 类似的也可以使用picard
java -jar picard.jar MarkDuplicates \
I=E_coli_K12.sorted.bam \
O=E_coli_K12.sorted.markdup.bam \
M=E_coli_K12.sorted.markdup_metrics.txt
picard 也提供了直接删除重复序列的操作。
直接添加额外选项,设定为true 即可REMOVE_DUPLICATES=true。
创建序列索引
这一步的目的,是方便我们可以快速地访问基因组上任意位置的比对情况,有助于我们随时了解数据。
samtools index E_coli_K12.sorted.markdup.bam
局部重比对
- 这一步与下面的BQSR 在gatk 分析E.coli K12 的本次实战中都没有应用。原因有二。
没有局部重比对是因为GATK 4.0 中没有相应的功能项,但GATK4.0 也提供了GATK HaplotypeCaller这个功能实现同样的结果。
而没有BQSR 则是因为E.coli K12没有那些必须的已知的变异集。
但我还是来了解一下重比对及BQSR 的执行过程及它们的意义。

开始前的合并
由上图所显示的那样,一般在局部重比对开始前,还有一部merge操作。它的作用是将同个样本的所有比对结果合并为一个大的bam文件。可以借助samtools 实现。
一般来说有些样本测序深度会很高,其测序结果需要经过多次测序(或者分布在不同的测序lane中)才能全部获得。
这时候一般是先比对排序并去重后再进行合并。
samtools merge [ ... ]
开始局部重比对
局部重比对的目的是将BWA 比对过程中所发现有潜在序列插入或者序列删除(insertion和deletion,简称Indel)的区域进行重新校正。这个过程往往还会把一些已知的Indel区域一并作为重比对的区域。
那么为什么要进行校正这一步呢?
原因就在于参考基因组的序列特点以及bwa、bowtie 这类序列比对算法本身。这类在全局搜索序列最优匹配的算法在存在Indel 区域的及其附近时往往会不准确。【特别是当一些存在长Indel、重复性序列的区域或者存在长串单一碱基(比如,一长串的TTTT或者AAAAA等)的区域中更是如此。】
另外,在这些比对算法中,对于碱基错配以及出现间隙的容忍度即罚分情况是不同的,而一般的序列比对算法则更倾向于碱基错配的情况。这就会导致基因组上本来是非常大的一段Indel 的地方,被错误的变为错配内容与短的gap。(关于双序列比对罚分规则的影响,可以参见:https://www.yuque.com/mugpeng/bioinfo/fgk5gz#b95f85b7
这必然会让我们检测到很多错误的变异。而且,这种情况还会随着所比对的read长度的增长(比如三代测序的Read,通常都有几十kbp)而变得越加严重。
而进行序列重比对,就使用了Smith-Waterman 算法,实现对全局比对结果的校正和调整,最大程度低地降低由全局比对算法的不足而带来的错误。而且GATK的局部重比对模块,除了应用这个算法之外,还会对这个区域中的read进行一次局部组装,把它们连接成为长度更大的序列,这样能够更进一步提高局部重比对的准确性。
比对先后的不同。(白色为空位,黑色为读取序列,彩色为错配)

GATK 提供了相关的功能。(推测这里作者使用的是3.x 版本的GATK)
- 首先是通过
RealignerTargetCreator,定位出所有需要进行序列重比对的目标区域。

java -jar GenomeAnalysisTK.jar \
-T RealignerTargetCreator \
-R human.fasta \
-I sample_name.sorted.markdup.bam \
-known 1000G_phase1.indels.b37.vcf \
-known Mills_and_1000G_gold_standard.indels.b37.vcf \
-o sample_name.IndelRealigner.intervals
- 接着通过
IndelRealigner对所有在第一步中找到的目标区域运用算法进行序列重比对,最后得到新的结果。也就是上图的after 的结果。
java -jar GenomeAnalysisTK.jar \
-T IndelRealigner \
-R human.fasta \
-I sample_name.sorted.markdup.bam \
-known 1000G_phase1.indels.b37.vcf \
-known Mills_and_1000G_gold_standard.indels.b37.vcf \
-o sample_name.sorted.markdup.realign.bam \
--targetIntervals sample_name.IndelRealigner.intervals
需要特别说明的是,-R 参数所用的参考序列为.fasta 格式。
这里还需要额外说明VCF文件。1000G_phase1.indels.b37.vcf, Mills_and_1000G_gold_standard.indels.b37.vcf分别来自于千人基因组项目和Mills项目,记录了那些项目中检测到的人群Indel区域。
候选的重比对,除了要在样本自身的比对结果中寻找外,还应该把人群已知的Indel 区域也包含进来。这些文件可以在GATK bundle ftp 中下载,(ftp://ftp.broadinstitute.org/bundle/)该版本需和参考基因组版本一致。
当然我们之前也说过了,如果我们在后面的变异检测使用GATK 的GATK HaplotypeCaller 模块,则可以减少这个局部重比对过程。因为GATK HaplotypeCaller 模块就包括了对潜在的变异区域进行相同的局部重比。
碱基质量重校正 BQSR
之前提到过,在上面的E_coli_K12基因组分析过程里,由于没有已知的变异集,无法进行这一步的操作。但这并不代表它不重要。
碱基质量值是衡量测序出来的碱基准确性的重要指标,而变异检测也是极度依赖碱基质量值。
而测定的碱基质量值与真实结果(实际的碱基质量)之间的偏差大小,也受到多方因素的影响。包括物理和化学等对测序反应的影响,甚至连仪器本身和周围环境都是其重要的影响因素。
BQSR(Base Quality Score Recalibration)这一方法也就应运而生。其主要采用机器学习的方法来构建测序碱基的错误率模型,并对这些碱基的质量值进行相应的调整。
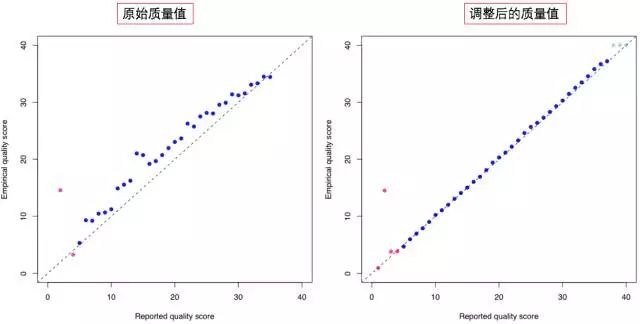
通过对比不难发现,经过BQSR 校正后,横轴的理论值 更加接近了纵轴真实值的数值,图像也拟合为了一条直线。
但是,理论值我们可以通过fastq 文件获得,真实值是如何产生的?
"真实值"是采用统计学的方法,获得极其接近的分布结果。下面是寻求“真实值”的思路。
假如一个碱基的质量报告为
20。根据公式Q = -10log(p_error),则错误率也就是10**-2 为1%。换句话说,意思就是如果有100个碱基的质量值均为20,则其中将会有一个碱基测定结果是错的。现在的问题就转换成了如何找到这些错误的碱基。
通常来说人与人之间基因的整体差异是很小的,那么一般来说在一个群体中发现的已知变异,在某个人身上也很可能是同样存在的。
因此我们可以尝试排除掉所有的已知变异位点,接着计算每个报告中(测序结果)的质量值和参考基因组比对有多少是不同的,这些不同的就可以认定为是错误的碱基,这些不同的碱基的数目所占整体其所属的质量值下相同的碱基的总数目的比例,就是真实的碱基错误率。再通过
Q = -10log(p_error)换算成质量分数,就可以和测得的质量数进行比较了。不难发现,在上图的原始质量值中,大部分的质量值都被高估了。
和局部重比对一样,BQSR 也包含了两个步骤。
1)通过BaseRecalibrator,计算出所有需要进行重校正的read和特征值,并将这些信息输出为一份校准表文件(sample_name.recal_data.table)。
java -jar GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R human.fasta \
-I sample_name.sorted.markdup.realign.bam \
—knownSites 1000G_phase1.indels.b37.vcf \
—knownSites Mills_and_1000G_gold_standard.indels.b37.vcf \
—knownSites dbsnp_138.b37.vcf \
-o sample_name.recal_data.table
2)然后通过PrintReads,利用第一步得到的校准表文件(sample_name.recal_data.table)重新调整来BAM文件中的碱基质量值,并输出一份新的BAM文件。
java -jar /path/to/GenomeAnalysisTK.jar \
-T PrintReads \
-R human.fasta \
-I sample_name.sorted.markdup.realign.bam \
—BQSR sample_name.recal_data.table \
-o sample_name.sorted.markdup.realign.BQSR.bam
这里必须注意,BQSR实际是为了校正测序过程中的系统性错误,因此,在执行的时候也是按照不同的测序文库进行处理。而这时候@RG信息就显得尤为重要了,BQSR 就是通过识别@RG中的ID信息,从而分辨各个测序过程。
变异检测
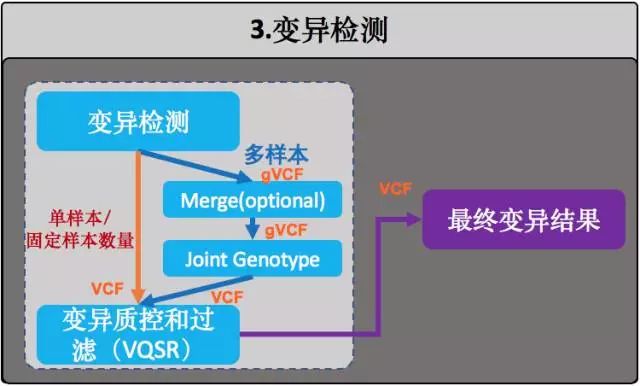
获得最终的变异结果,是一般的全基因组分析的流程的目标。
变异检测的内容一般会包括:SNP、Indel,CNV和SV等。而这里使用GATK HaplotypeCaller模块对样本中的变异进行检测,它也是目前最适合用于对二倍体基因组进行变异(SNP+Indel)检测的算法。
HaplotypeCaller 与一般的直接应用贝叶斯推断的算法有所不同,它会先推断群体的单倍体组合情况,计算各个组合的几率,然后根据这些信息再反推每个样本的基因型组合。因此它不但特别适合应用到群体的变异检测中,而且还能够依据群体的信息更好地计算每个个体的变异数据和它们的基因型组合。
如图上所示,变异检测应用HaplotypeCaller有两种做法。对应于但样本与多样本的情况。
做法一
直接使用HaplotypeCaller。这个方法适合单样本,或者固定样本的情况。但缺陷是,如果需要增加新的样本就必须要重新运行HaplotypeCaller,也就是所谓N+1 难题。
java -jar /path/to/GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R /path/to/human.fasta \
-I sample_name.sorted.markdup.realign.BQSR.bam \
-D /path/to/gatk/bundle/dbsnp_138.b37.vcf \
-stand_call_conf 50 \
-A QualByDepth \
-A RMSMappingQuality \
-A MappingQualityRankSumTest \
-A ReadPosRankSumTest \
-A FisherStrand \
-A StrandOddsRatio \
-A Coverage
-o sample_name.HC.vcf
- 其中
-D参数输入的dbSNP 也同样可以在GATK bundle 中找到,这份文件汇集的是目前几乎所有的公开人群变异数据集。
除外,如果我们有多个.bam样本输入则可以继续使用-I参数输入。-I sample1.bam [-I sample2.bam ...]
一般来说,若是直接对人类基因组做变异检测,由于人类基因组数据过于庞大,往往整个过程会消耗大量的时间,通常需要几十个小时甚至几天。因此我们为了提高效率可以采用分布执行的方法。
我们可以将基因上不同的染色体理解为相互独立的(结构性变异除外),也就是说,我们可以对待不同的染色体,一条条独立的来执行变异检测这个步骤,最后再把结果合并起来。这里就额外指定一个参数-L, 一般直接写-L 1,但有的也会书写-L chr1,这是由fasta 文件中的命名决定的。
做法二
第二个方法是在第一个方法之上,先将每个样本各自生成一个gVCF,再对整个群体进行joint-genotype。
gVCF 全称为genome VCF,是每个样本用于变异检测的中间文件,格式类似于VCF,它将整个joint-genotype 过程中需要的所有信息都记录在这里面。而从文件大小上和数据量上,都远远小于原来的BAM文件。这样一旦新增加样本也不需要再重新去读取所有人的BAM文件,只需要额外生成一份gVCF 并再进行一次joint-genotype就好。
开始变异检测
在开始之前可以利用CreateSequenceDictonary 功能为E.coli K12的参考序列生成一个.dict 的文件。
gatk CreateSequenceDictionary -R E.coli_K12_MG1655.fa \
-O E.coli_K12_MG1655.dict
接着就开始变异检测了。这里我们使用上面提到的做法二。
- 首先需要生成gVCF 的中间文件。
gatk HaplotypeCaller -R E.coli_K12_MG1655.fa \
> --emit-ref-confidence GVCF \
> -I ~/20200524_wgs_practice/output/E.col/E_coli_K12.sorted.markdup.bam \
> -O ~/20200524_wgs_practice/output/E.col/E_coli_K12.g.vcf
这个过程一般会持续比较久的时间,比如仅仅是E.coli_K12,就耗费了Elapsed time: 3.79 minutes。
其中--emit-ref-confidence GVCF 表示产生gVCF 的指令。
- 接着通过生成的gVCF检测变异
gatk GenotypeGVCFs \
> -R ~/20200524_wgs_practice/input/E.col/fasta/E.coli_K12_MG1655.fa \
> -V E_coli_K12.g.vcf \
> -O E_coli_K12.vcf
这样我们就获得了E.coli K12 样本的变异结果E_coli_K12.vcf。
接着我们就可以用tabix为它构建索引,方便以后的分析。
tabix -p vcf
提示需要先将其压缩。
[tabix] was bgzip used to compress this file? E_coli_K12.vcf
压缩一下
bgzip -f E_coli_K12.vcf
再重新执行tabix 即可。
变异检测质控和过滤(VQSR)
和原始数据的质控一样,对变异检测的结果进行质控和过滤,也是同样的重要。VQSR,Variant Quality Score Recalibration,是通过构建GMM模型对好和坏的变异进行区分,从而实现对变异的质控。它通过机器学习的方式,利用多个不同的数据特征训练一个模型以对变异数据进行质控。使用VQSR 需要具备两个条件。
条件1:已知变异集
首先需要一个已知的变异集,将作为训练质控模型的数集。现在有Hapmap、OMNI,1000G和dbsnp等这些国际性项目的数据,可以作为高质量的变异集。GATK的bundle主要就是对这四个数据集做了精心的处理和选择,然后把它们作为VQSR时的真集位点。
需要注意的是,VQSR并不是用这些变异集里的数据来训练的,而是用我们自己的变异数据。这些国际项目的数据集只是为我们提供了位点,告诉模型群体中哪些位点存在着变异,如果在其他人的数据里能观察到落入这个集合中的变异位点,那么这些被已知集包括的变异就有很大的可能是正确的。
因此我们可以从数据中筛选出那些和国际项目的数据集 也就是真集相同的变异,并把它们当作真实的变异。接着,进行VQSR的时候,程序就可以用这个筛选出来的数据作为真集数据来训练,并构造模型了。
VQSR 的原理
VQSR 的核心就在于构建一个区分“好”与“坏”变异的分类器。
该分类器是通过GMM 模型来构建的。这个模型在构造时,并不是使用所有的数据来进行构造的。而是挑出一部分,那些可以和已知的变异集合位点(国际项目的参考数据集,Hapmap、千人数据等)对应的数据,为它们分配不同的高可信度权重。基于群体遗传的原理,已知的变异会被认为是更加靠谱的变异,因此在初始化的时候先把它们当作是正确的变异。
利用这个初始变异集 ,我们可以按照特征训练一个区分好变异的GMM 模型,接着对全部数据进行打分,再把那些评分低的挑选出来,构成一个最不正确的变异集合,并用这个集合构建一个区分坏变异的GMM。
最后再同时利用好变异与坏变异的GMM 对变异数据进行打分,通过该变异的评分,就可以评判这个变异的质量值了。
条件2: 检测结果有足够多的变异
检测结果必须有足够多的变异信息,不然VQSR在进行模型训练的时候会因为可用的变异位点数目不足而无法进行。
由于条件1的限制(已知的变异集),包括我们上面的E.coli K12 在内,很多的非人的物种在完成变异检测后是无法进行VQSR 的。
而条件2则会导致某些捕获测序(Panel测序数据,甚至外显子测序)的数据,由于变异位点的不够,也无法只使用VQSR。
硬过滤是什么
当无法使用VQSR时,就不得不考虑硬过滤的方法了。
所谓硬过滤,就是人为的设定一个或者若干个指标阈值(数据特征值),然后把所有不满足阈值的变异位点采用一刀切的方法切除。
我们该如何执行硬过滤呢?或者说,该如何选择指标用以标准来评定变异呢?
我们可以直接使用gatk_vqsr 中精心挑选的指标。这些指标一共有6个(这些指标都在VCF文件的INFO域中,如果不是GATK得到的变异,可能会有所不同,但知道它们的含义之后我们也是可以通过自己计算来确定的)
包括:
1.QualByDepth(QD)
2.FisherStrand (FS)
3.StrandOddsRatio (SOR)
4.RMSMappingQuality (MQ)
5.MappingQualityRankSumTest (MQRankSum)
6.ReadPosRankSumTest (ReadPosRankSum)
那么我们该如何设定这些阈值呢?
QualByDepth(QD)
QD 是变异质量值(Quality)除以覆盖深度(Depth)得到的比值。而这里的变异质量值就是VCF中QUAL的值。QUAL 用来用来衡量变异的可靠程度;而Depth则表示这个位点上所有的含有变异碱基的样本的覆盖深度之和。通俗来说,这个数值可以由每个含变异的样本的覆盖深度累加获得。
其中,对于样本中的G/T 非0 样本(GT=1/1表示纯合变异;GT=0/1表示杂合变异;GT=0/0表示非变异),计算它们的加和。并按照定义,QD值即为质量值QUAL 除以含有变异样本的深度之和Depth。
QD这个值描述的实际上就是单位深度的变异质量值,一般来说QD 越高变异的可信度也就越高。
但是QD 的数值是如何确定的呢?可以通过VQSR质控先后的数据来进行解释。(这里选用的是NA12878 的数据)
我们可以先把所有变异位点的QD值都提取出来,然后画一个密度分布图。通过这个分布图,不难了解到,QD的范围主要集中在0~40之间。而两个峰值分别为QD = 12 以及QD = 32。
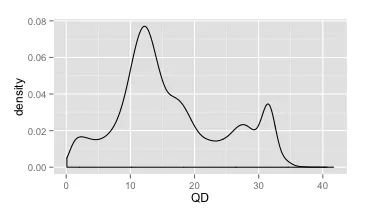
其中第一个峰(QD=12)代表杂合,而第二峰(QD=32)代表纯合。因为对于纯合变异来说,贡献给质量值的read 是杂合变异的两倍,因而同样深度的情况下,QD 会更大一些。
接着我们可以画出VQSR 后的所有可信变异(绿色),与不可信变异(红色)的QD分布图。
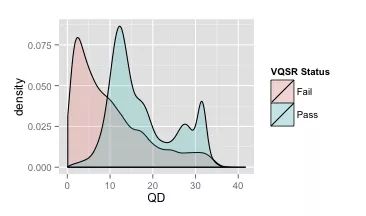
由图可见,大部分的不可信变异都集中在了左侧的低QD区域,而恰好红色区域的波峰为QD = 2 处。
但我们也不难发现,在高的QD 区域其实也存在小部分的不可信变异(红)分布;而同样在低QD 区域也存在小部分的可信变异(绿)。而这样的结果也是硬过滤的弊端。它不会像VQSR那样,通过多个不同维度的数据构造合适的高维分类模型,从而能够更准确地区分好和坏的变异,而仅仅是一刀切。
FisherStrand (FS)
FS 是通过Fisher检验的p-value转换而来的值,它是用来描述测序或者比对时对于只含有变异的read以及只含有参考序列碱基的read是否存在着明显的正负链特异性(Strand bias,或者说是差异性)
这个差异反映了测序过程是否随机,或者说比对算法是否在基因组的某些区域存在一定的选择倾向。如果测序过程是随机的,而且比对不存在问题,则无论read 是否存在变异,再或者该变异来自于基因组的正链或负链,只要是真实的,这些read 都应该是比较均匀的,也就是说FS 的值应该接近于零,即不出现链特异的比对结果。
除此之外,在VCF文件的INFO区域内,有些时候除了FS,还可能会有SB 或SOR。它们实际上也是从不同层面对链特异描述的指证。SB 表示原始各链比对数目,而FS 是对结果进行了Fisher检验后转换而来的结果;SOR则和FS 类似,只是使用了不同的统计方法来计算差异程度,它更适合高覆盖度的数据。
而FS 硬过滤数值的确定,和QD 的确定过程是类似的。
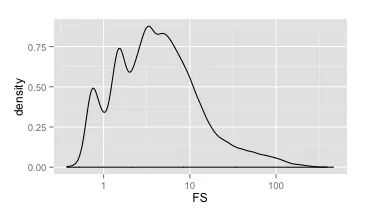
由图可见,FS 的数值范围非常广泛,而大多数的结果还是集中在了100以内。

而经过VQSR 后的结果,和QD 的图也是类似的。所有可信变异(绿色),与不可信变异(红色)。大部分的可信变异都在
0-10之间,而大部分的不可信变异都集中在60附近。因此过滤的时候,我们把FS设置为大于60。虽然这么做看起来很激进(60以下还是有很多的不可信变异红色区域),但是由于不可信变异的变化非常平缓,无论如何都会保留大部分,倒不如选择一个可以保留多的可信变异内容的数值,再借助其他的过滤指标进行筛选。
StrandOddsRatio(SOR)
和FS 一样,SOR 也是对链特异的一种描述。由于常常FS 在硬过滤的时候不是很能满足过滤的需求(如上所述的那样),而且很多时候read 在外显子区域末端的覆盖存在着一定的链特异(是正常的),往往只有一个方向的read,这个时候如果该区域中有变异位点的话,则FS 会给出很差的分值,而SOR 就可以起到补充校正的作用了。
SOR 计算使用的是symmetric odds ratio test,数据表现为一个2×2的列联表。

考虑的其实就是ALT和REF这两个碱基的read覆盖方向的比例是否有偏,如果完全无偏,那么应该等于1。
SOR = (ref_fwd/ref_rev) / (alt_fwd/alt_rev)
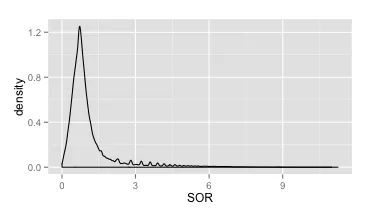
可以看到,大部分的SOR 分布集中在0~3 之间。

可信变异大部分集中在0~3 之间,因此可以将阈值选择为3。
RMSMappingQuality(MQ)
MQ 表示的是所有比对至该位点上的read的比对质量值的平方和的平均值的平方根。

这个数值相比平均值,能够更好的反应整体的比对质量值的离散程度。如果我们的比对工具用的是bwa mem,那么按照它的算法,对于一个好的变异位点,我们可以预期,它的MQ值将等于60。
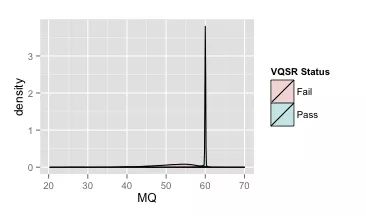
由图显示,在40以下,不可信变异几乎完全没有了,因此可以将阈值设定为40。
除此之外,对于MappingQualityRankSumTest(MQRankSum) 与ReadPOSRankSumTest(ReadPOSRankSum)的设定原理,和其他四个是完全一样的, 都是通过VQSR 密度分布图,确定了硬过滤的阈值(保留大多数的可信变异,剔除大多数的不可信变异)。
执行硬过滤
gatk 提供了VariantFiltration 模块来进行硬过滤。但需要注意的是,过滤的时候需要分SNP 与Indel 两个不同的变异类型来进行。因为这两个变异类型的一些阈值是不同的,需要区别对待。
为SNP进行硬过滤
首先要选择出SNP
gatk SelectVariants \
-select-type SNP \
-V ../output/E.coli/E_coli_K12.vcf.gz \
-O ../output/E.coli/E_coli_K12.snp.vcf.gz
接着对这些SNP 进行硬过滤。
gatk VariantFiltration \
-V ../output/E.coli/E_coli_K12.snp.vcf.gz \
--filter-expression "QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \
--filter-name "PASS" \
-O ../output/E.coli/E_coli_K12.snp.filter.vcf.gz
为Indel 进行硬过滤
选择出Indel
gatk SelectVariants \
-select-type INDEL \
-V ../output/E.coli/E_coli_K12.vcf.gz \
-O ../output/E.coli/E_coli_K12.indel.vcf.gz
对Indel 进行过滤
gatk VariantFiltration \
-V ../output/E.coli/E_coli_K12.indel.vcf.gz \
--filter-expression "QD < 2.0 || FS > 200.0 || SOR > 10.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \
--filter-name "PASS" \
-O ../output/E.coli/E_coli_K12.indel.filter.vcf.gz
合并过滤结果
上面关于Indel 与SNP 的过滤所使用的阈值都是借用NA12878(来自GIAB)的高深度数据进行计算获得的,但如果序列来源不是哺乳动物,就最好选定新的合适的阈值。
gatk MergeVcfs \
-I ../output/E.coli/E_coli_K12.snp.filter.vcf.gz \
-I ../output/E.coli/E_coli_K12.indel.filter.vcf.gz \
-O ../output/E.coli/E_coli_K12.filter.vcf.gz
Ti/Tv 的范围
可以将Ti/Tv 值理解为物种在与自然相互作用和演化过程中在基因组上留下来的一个统计标记,在物种中这个值具备一定的稳定性。
Ti 是转换 Transition, 指的是嘌呤转嘌呤,嘧啶转嘧啶,A-G,C-T。
Tv是颠换 Transversion,指的是嘌呤转嘧啶,嘧啶转嘌呤,A-C,A-T,G-T或 G-C。
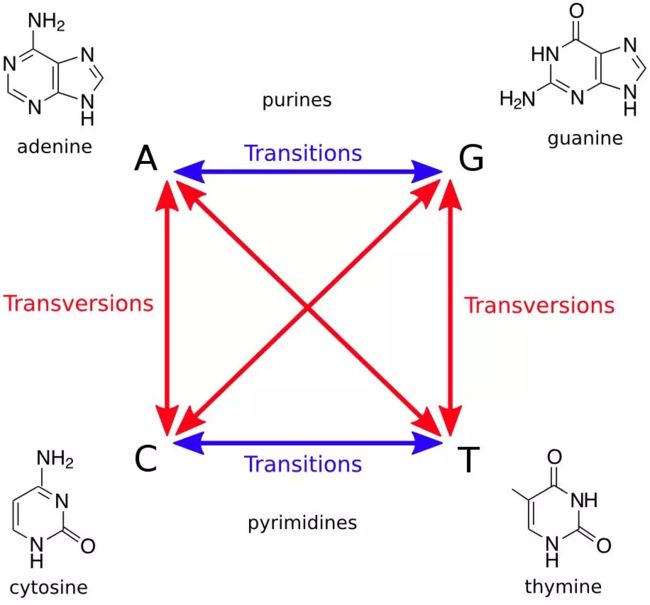
因此在完成变异质控之后,我们最好再看一下这些变异位点Ti/Tv的值是多少,以此来进一步确定结果的可靠程度。
另外,在哺乳动物基因组上C->T的转换比较多,这是因为基因组上的胞嘧啶C在甲基化的修饰下容易发生C->T的转变。
一般来说,如果没有选择压力,Ti/Tv将等于0.5,因为从概率上来说,Tv 发生的概率是Ti 的两倍。而事实上,对于不同的物种,有不同的差异。对于人来说,全基因组正常的Ti/Tv在2.1左右,而外显子中则在3.0,新发生的变异则在1.5左右。(Tv 更容易发生)
Ti/Tv 实际上是一个被动指标,因此我们只能够用它来检验结果的可靠程度,而不能够将其作为一个变异质控的过滤标准。
最后
其实我们都可以把各个步骤的命令写称shell 脚本保存起来,这样就像程序一样,我们可以合并或者修改脚本中的某些参数,达到一个快速的流程化的全基因组分析。