测序上游分析系列:
mRNA-seq转录组二代测序从raw reads到表达矩阵:上中游分析pipeline
miRNA-seq小RNA高通量测序pipeline:从raw reads,鉴定已知miRNA-预测新miRNA,到表达矩阵【一】
miRNA-seq小RNA高通量测序pipeline:从raw reads,鉴定已知miRNA-预测新miRNA,到表达矩阵【二】
其他文章系列:ggplot2作图篇:
(1)基于ggplot2的RNA-seq转录组可视化:总述和分文目录
(2)测序结果概览:基因表达量rank瀑布图,高密度表达相关性散点图,注释特定基因及errorbar的表达相关性散点图绘制
(3)单/多个基因在组间同时展示的多种选择:分组小提琴图、分组/分面柱状图、单基因蜂群点图拼图的绘制
(4)配对样本基因表达趋势:优化前后的散点连线图+拼图绘制
TCGA转录组数据在完成差异分析后,我们通常希望系统地获取这些成百上千的差异基因的功能信息,帮助我们分析下游实验的思路。面对大量的差异基因,逐个查询基因功能是不切实际的。所以我们需要借助基因功能富集分析工具,获取有意义的功能基因集。
基因功能富集分析的基本思路是一致的:开发者将具有相似功能的基因划分到同一个基因集中,然后使用统计检验分析测序获得的差异基因是否显著聚于这些已知功能的基因集中。若检验结果显著,则可说明差异基因可能参与了这一基因集所注释的生物功能,为我们的下游研究提供了系统精准的思路。目前常用的基因富集分析方法有超几何检验富集分析、Gene Set Enrichment Analysis (GSEA)和Gene Set Variation Analysis (GSVA) (或功能类似的single sample Gene Set Enrichment Analysis (ssGSEA))。
如何从TCGA数据库中获得差异基因?我们从这里开始:
- 从TCGA数据库下载并整合清洗高通量肿瘤表达谱-临床性状数据
- TCGA数据整合后进行DESeq2差异表达分析和基于R的多种可视化
下面使用的对象名称和内容与前文保持一致,仍使用之前教程选择的的TCGA-LUSC白色人种肺鳞癌表达谱数据(整合后cancer=344, normal=42)。
进行基因富集分析,我们需要的R对象是:
(1)dds_DE: this object created by DESeq2 contains results of differential expression analysis. We need to extract log2FoldChange, gene symbol and other available information from it.
(2)condition_table: this table contains the grouping info of all samples. It will be used to create a new grouping table compatible with Limma package.
(3)mRNA_exprSet: the expression matrix containing columns 'gene_name' and 'gene_id' representing GENE SYMBOL and ENSEMBL ID. They will be used for ID transfer.
注:这里的mRNA_exprSet相比第一篇教程整合得到的矩阵多了一行'gene_id'。因为考虑到基因的各种ID之间并没有非常完美的对应,尤其是SYMBOL(基因的各种名字版本更新或许不齐),所以既然在整合的时候已经进行过一次SYMBOL-ENSEMBL ID转换,尽可能还是用相同批次的数据。其实差别不大:
#the last filtering step I used in the tutorial before:
mRNA_exprSet <- mRNA_exprSet[,c('gene_name', condition_table$TCGA_IDs)]
#We just don't need to remove 'gene_id' column:
mRNA_exprSet <- mRNA_exprSet[,c('gene_name', 'gene_id', condition_table$TCGA_IDs)]
1. 超几何检验GO、KEGG基因富集分析
这是相对简单粗暴一些的基因富集分析方法。不需要输入基因的表达值,只需要通过DESeq2和自己设定的阈值(如|log2FoldChange| > 2 & FDR < 0.05)筛选得到的差异基因名。进行统计检验后返回显著富集的功能基因集。
GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)是两种常用的数据库。GO中的功能数据集分为3个sub-classes: Biological Process (BP), Molecular Function (MF) 和 Cellular Component (CC),分别含有生物学功能活动、分子功能和细胞组分定位的基因集合(GO terms)。KEGG包含的基因集合均为在某特定生物学功能过程中发挥作用的分子信号通路(KEGG pathways)。
(1)需要R包。
DESeq2 (获得差异基因信息), clusterProfiler(ID转换+富集分析+作图一站式神包!)。
install.packages('DESeq2')
source("https://bioconductor.org/biocLite.R")
biocLite("clusterProfiler") #this package can be downloaded from Bioconductor.
library(DESeq2)
library(clusterProfiler)
(2)提取差异基因列表。
根据我们指定的硬阈值提取出差异表达基因的基因SYMBOL(做差异分析时输入的矩阵中rownames为SYMBOL)。
res_DE <- results(dds_DE, alpha=0.05, contrast=c("sample_type","cancer","normal"))
resSig <- subset(res_DE,padj < 0.05)
resSigAll <- subset(resSig, (log2FoldChange < (-2)|log2FoldChange > 2))
gene_list_for_GO_KEGG <- resSigAll@rownames
我们一般会遇到的基因ID类型有:ENSEMBL GENE ID (如ENSG00000186092),GENE SYMBOL(如OR4F5)和ENTREZID(如79501)。绝大部分的基因ID都是能够一一对应的,但自然是不排除有例外...
clusterProfiler包的KEGG分析函数只接受基因的ENTREZID而不是SYMBOL,所以我们进行基因ID的转换。而且一般由于SYMBOL版本繁杂,也建议用ENTREZID进行富集分析比较稳妥,防止过多SYMBOL不能和数据库匹配而导致分析结果的不可信。ENSEMBL和SYMBOL之间的转换可以直接借助第一篇数据整合教程中的gtf文件来完成。但其中不含对于富集分析较为关键的ENTREZID。这里我们使用bitr函数可以进行SYMBOL, ENSEMBL ID和ENTREZID的在线转换。
可想而知,编号之间的对应关系更好。所以我习惯用ENSEMBL ID和ENTREZID互换。这里比较懒,普通的GO/KEGG分析就直接用SYMBOL转换了,只是可能转换丢失率会稍高一些。但丢失非常少部分的基因是合理而且问题不大的。
#from/toType = 'SYMBOL'/'ENTREZID'/'ENSEMBL'
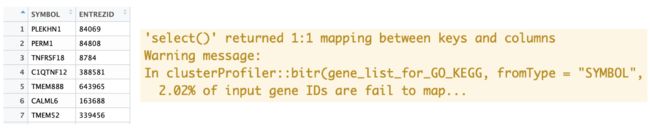
gene_list_for_GO_KEGG <- clusterProfiler::bitr(gene_list_for_GO_KEGG, fromType = 'SYMBOL',
toType = 'ENTREZID', OrgDb = 'org.Hs.eg.db')
于是便获得了具有对应信息的table和提示有基因转换丢失Warning message:
(3)GO单种subclass的富集分析。
接下来就可以用enrichGO函数分析和可视化了。先只做Biological Process亚类的分析。(还是懒,就直接用SYMBOL做GO分析了)
注:函数keyType指输入的ID类型,OrgDb为数据库种属,ont为subclass类型(BP/CC/MF/All)
GO_BP <- enrichGO(gene = gene_list_for_GO_KEGG$SYMBOL, keyType = 'SYMBOL', OrgDb = 'org.Hs.eg.db', ont = 'BP', pvalueCutoff = 0.05)
barplot(GO_BP, showCategory = 20, title = 'Top20 GO-BP terms', x = 'Count', color = 'p.adjust') #ont='BP'/'MF'/'CC', x='Count'/'GeneRatio'
dotplot(GO_BP, title = 'Top20 GO-BP terms', showCategory = 20, color = 'p.adjust')

结果解释:一般可视化top10/20的富集基因集合。barplot x轴设定为在该基因集合中的enriched gene counts,颜色代表FDR,排序根据FDR。dotplot横轴设定GeneRatio = m/n(m为在某显著子基因集中的输入差异基因个数,n为能在总BP基因集中检测到的输入差异基因数),气泡大小为绝对enriched gene counts数,颜色代表FDR,排序是根据gene ratio。个人比较喜欢气泡图,多一个维度的数据,而且排序根据富集程度而不是FDR显著程度。(类比一下普通RT-PCR实验:我们通常只需要p值显著<0.05,相比之下更关心的绝对是fold change)
不过本人认为clusterProfiler包dotplot默认的x轴排序方法GeneRatio仍旧不是最好的办法,以enrichment score(存在于某子基因集中的输入差异基因/该子基因集的总基因数)或许比较客观,之后可以使用ggplot2自己实现一下。
对GO三个subclasses的基因集合同时做富集分析:
GO_all <- enrichGO(gene = gene_list_for_GO_KEGG$SYMBOL,ont = 'All' , keyType = 'SYMBOL', OrgDb = 'org.Hs.eg.db', pvalueCutoff = 0.05)
dotplot(GO_all, title = 'Top10 GO terms of each sub-class', showCategory = 10, color = 'p.adjust', split='ONTOLOGY')+ facet_grid(ONTOLOGY~.,scale="free")
可以获得分面的富集图,每个subclass选取Top10可视化:

(4)KEGG富集分析。
KEGG <- enrichKEGG(gene_list_for_GO_KEGG$ENTREZID, organism = 'hsa', pvalueCutoff = 0.05)
dotplot(KEGG, title = 'Top20 KEGG pathways', showCategory = 20, color = 'p.adjust')
品味一下KEGG的富集通路,是不是内味和GO略微不同呢~~

2. GSEA富集分析
相比于第一种简单粗暴的用硬阈值截取+往篮子里塞鸡蛋的方法,GSEA同时考虑了基因在整个表达谱中所处的FoldChange rank以及同一基因集中的基因在表达谱rank中的距离。通俗来讲,GSEA基于如下假设:一个基因集中的基因如果在表达谱中所处的rank越极端(高/低FoldChange),而且基因之间的距离越短(rank相近),则这个基因集越可能显著。所以GSEA需要输入的是【所有纳入差异分析的完整基因list和它们的FC值,并已经预先排序(pre-ranked)】。
(1)需要R包:
DESeq2 (获得差异基因信息),clusterProfiler(ID转换+富集分析+作图一站式神包!),dplyr(表格操作),ggplot2(修饰clusterProfiler的作图),pheatmap(热图绘制)。
install.packages('dplyr)
install.packages('DESeq2')
install.packages('ggplot2')
install.packages('pheatmap')
source("https://bioconductor.org/biocLite.R")
biocLite("clusterProfiler") #this package can be downloaded from Bioconductor.
library(DESeq2)
library(clusterProfiler)
library(dplyr)
library(ggplot2)
library(pheatmap)
(2)获取(几乎)所有基因的log2FC数据并进行ID转换。
首先使用results函数获得所有纳入差异分析的基因信息。(这函数一定得设置0-1间的阳性错误率,那就多打几个9吧,尽可能把完整基因谱都提取出来)
GSEA还是用ENTREZID好好做,所以祭出了之前用到的ENSEMBL ID和SYMBOL互换的mRNA_exprSet[,c(1,2)],至少可以保证SYMBOL到ENSEMBL ID的百分之百转换率。随后使用bitr将ENSEMBL对应到ENTREZID,去除重复后最终与log2FC数据合并。
dds_for_GSEA <- results(dds_DE, contrast=c("sample_type","cancer","normal"), alpha = (0.9999999999999999))
lFC_for_GSEA <- data.frame('gene_name'=dds_for_GSEA@rownames, 'lFC'=dds_for_GSEA$log2FoldChange, stringsAsFactors = F)
ENS_for_GSEA <- dplyr::filter(mRNA_exprSet, gene_name %in% dds_for_GSEA@rownames)[,c('gene_name','gene_id')] %>%
dplyr::inner_join(lFC_for_GSEA, by='gene_name')
ENTREZ <- clusterProfiler::bitr(ENS_for_GSEA$gene_id, fromType = 'ENSEMBL', toType = c('ENSEMBL','ENTREZID'), OrgDb = 'org.Hs.eg.db')
names(ENTREZ) <- c('gene_id', 'ENTREZID')
ENTREZ <- ENTREZ[!duplicated(ENTREZ$gene_id),]
ENTREZ <- ENTREZ[!duplicated(ENTREZ$ENTREZID),] %>% dplyr::inner_join(ENS_for_GSEA, 'gene_id')
(3)建立输入对象并分析表达谱在GO BP和KEGG中的富集程度。
提取对应上ENTREZID的log2FC vector并以ID作为vector各个元素的名字。按lFC降序排序后输入到专门用于GO数据库GSEA分析的gseGO函数,和专用于KEGG数据库GSEA分析的gseKEGG函数中进行分析。
注:nPerm指permutation重复检验富集是否随机的次数,建议500,1000或更多。keyType中’ncbi-geneid‘就是指ENTREZID。
input_GSEA <- ENTREZ$lFC
names(input_GSEA) <- ENTREZ$ENTREZID
input_GSEA <- sort(input_GSEA, decreasing = T)
GSEGO_BP <- gseGO(input_GSEA, ont = 'BP', OrgDb = 'org.Hs.eg.db', nPerm = 1000, pvalueCutoff = 0.05)
GSEA_KEGG <- gseKEGG(input_GSEA, organism = 'hsa', keyType = 'ncbi-geneid', nPerm = 1000, pvalueCutoff = 0.05)
(4)GSEA结果的可视化和解读。
将分析获得的对象压缩到扁平的dataframe,判定富集基因集,并根据NES排序。
GSEA中判断基因集是否富集一般取决于如下参数:
(1)NES(normalized enrichment score): 绝对值>1为富集,严苛可>1.5。
(2)p值:<0.05,严苛可<0.01。
(3)q值:<0.25,严苛可<0.1/0.05/0.01。
找出有兴趣进行可视化的显著基因集,并使用gseaplot和gseaplot2分别可视化。(我这里用ggplot2稍微加了个字幕~)
#further filter out the significant gene sets and order them by NES scores.
#GO
GSEA_BP_df <- as.data.frame(GSEGO_BP) %>% dplyr::filter(abs(NES)>1 & pvalue<0.05 & qvalues<0.25)
GSEA_BP_df <- GSEA_BP_df[order(GSEA_BP_df$NES, decreasing = T),]
p <- gseaplot(GSEGO_BP, GSEA_BP_df$ID[1], by='all', title = GSEA_BP_df$Description[1], color.vline = 'gray50', color.line='red', color='black') #by='runningScore/preranked/all'
p <- p+annotate(geom = 'text', x = 0.87, y =0.85, color='red', fontface = 'bold', size= 4,
label=paste0('NES= ', round(GSEA_BP_df$NES[1], 1), '\n', 'p.adj= ', round(GSEA_BP_df$p.adjust[1], 2)))+
theme(panel.grid = element_line(colour = 'white'))
p
#KEGG
GSEA_KEGG_df <- as.data.frame(GSEA_KEGG) %>% dplyr::filter(abs(NES)>1 & pvalue<0.05 & qvalues<0.25)
GSEA_KEGG_df <- GSEA_KEGG_df[order(GSEA_KEGG_df$NES, decreasing = T),]
p <- enrichplot::gseaplot2(GSEA_KEGG, geneSetID = GSEA_KEGG_df$ID[1], pvalue_table = F, ES_geom = 'line')+
annotate('text', x = 0.87, y =0.85, color='red', fontface = 'bold', size= 4,
label=paste0('NES= ', round(GSEA_KEGG_df$NES[1], 1), '\n', 'p.adj= ', round(GSEA_KEGG_df$p.adjust[1], 2)))+
labs(title = GSEA_KEGG_df$Description[1])+theme(plot.title = element_text(hjust = 0.5))
p
两种风格任选其一即可。左图的富集结果是相当相当好了。基因集中大部分基因都聚集在表达谱的正顶端,且基因rank距离近,曲线呈明显的偏分布正峰。

(5)查看GSEA显著基因集中基因在样本中的具体表达情况(热图可视化)。
得到了显著的基因集,但最终我们的落脚点还是在具体基因上。所以或许需要对我们感兴趣的基因集中参与富集的基因的表达情况进行可视化。
这里仍用GO BP富集NES最高的keratinization演示。热图没有进行聚类,col按样本类别分类,row按log2FC降序排列,比较直观清晰。
#to visualize the expression trends of genes within a significant GSEA geneset.
ENTREZID_within_a_set <- unlist(strsplit(GSEA_BP_df$core_enrichment[1], split = '/', fixed = T))
symbols_within_a_set <- dplyr::inner_join(ENTREZ, data.frame('ENTREZID'=ENTREZID_within_a_set, stringsAsFactors = F), by='ENTREZID')[,c('gene_name','lFC')]
symbols_within_a_set <- symbols_within_a_set[order(symbols_within_a_set$lFC, decreasing = T),]
sub_expr_norm <- expr_norm[symbols_within_a_set$gene_name,]
annotation_col <- data.frame('Sample_type'=condition_table$sample_type)
rownames(annotation_col) <- condition_table$TCGA_IDs
pheatmap::pheatmap(t(scale(t(sub_expr_norm))), show_colnames = F, show_rownames = T,
annotation_col = annotation_col, cluster_cols = F, cluster_rows = F,
color = colorRampPalette(c("navy", "white", "firebrick3"))(100), fontsize = 4,
annotation_colors = list(Sample_type=c(cancer = 'orange', normal = 'blue')),
legend = T, legend_breaks = c(-2, 0, 2), breaks = unique(seq(-2, 2, length=100)),
lagend_labels = c('≤2','0','≥2'), main = paste0('Heatmap of enriched genes within ', GSEA_BP_df$Description[1]))

(6)自建基因集合进行GSEA分析。
前面使用了函数内置好的基因集合进行GSEA。如果我们需要自定义基因集合怎么办?GSEA函数可以胜任,它接受表达情况和自定义基因集作为输入,进行GSEA分析。
作为测试,我去GSEA官网Molecular Signatures Database下载了hallmark gene sets的entrez gene id基因集文件(.gmt)。
用文本编辑器打开.gmt文件,发现就是我们理解的基因集格式:一个功能描述+网站广告后,紧接所有包含在本功能集合中的基因ID。
而GSEA函数要求的database输入也是一定的:包含两列的表格:第一列为功能基因集的名称;第二列为基因ID。

所以接下来是geneset格式的建立。
original_gmt <- readLines('h.all.v7.0.entrez.gmt')
strsplit_name <- function(gmt.list_layer){
GSgenes <- as.character(unlist(strsplit(gmt.list_layer, split = '\t', fixed = T)))
data.frame('Genesets'=rep(GSgenes[1], (length(GSgenes)-2)), 'Genes'=GSgenes[c(-1,-2)], stringsAsFactors = F)
}
database_list <- lapply(original_gmt, strsplit_name)
db_for_GSEA <- do.call(rbind, database_list)
db_for_GSEA$Genesets <- as.character(db_for_GSEA$Genesets)
db_for_GSEA$Genes <- as.character(db_for_GSEA$Genes)
同前,分析并可视化。gseaplot2可以同时可视化多个genesets。不过感觉用处不是太大。
GSEA_test <- clusterProfiler::GSEA(input_GSEA, nPerm = 1000, pvalueCutoff = 0.05, TERM2GENE = db_for_GSEA)
GSEA_test_df <- as.data.frame(GSEA_test) %>% dplyr::filter(abs(NES)>1 & pvalue<0.05 & qvalues<0.25)
GSEA_test_df <- GSEA_test_df[order(GSEA_test_df$NES, decreasing = T),]
enrichplot::gseaplot2(GSEA_test, GSEA_test_df$Description[c(1:2, (length(GSEA_test_df$Description)-1):length(GSEA_test_df$Description))], color = c('red','orange','blue','navy'))

3. GSVA/ssGSEA分析
ssGSEA是为无重复的样本进行geneset enrichment analysis准备的,所以不同于上方以组别为单位(cancer vs normal)的GSEA分析,通过ssGSEA,每个样本都可以得到相应基因集的评分。GSVA的原理和作用类似,所以GSVA和ssGSEA被写入了同一个R包中,性能等同。
所以我们甚至可以对每个样本的pathway scores进行差异分析,从而获得组间差异表达的pathways。这又是另外一种船新体验了有没有~现在兴起的新玩法还包括使用ssGSEA对肿瘤测序结果进行免疫细胞特征基因的评分,从而鉴定肿瘤免疫浸润的程度和特征。
(1)需要R包:
GSVA包(进行GSVA/ssGSEA分析),limma包(差异pathway的筛选),pheatmap包(热图绘制)。
source("https://bioconductor.org/biocLite.R")
biocLite(c("GSVA",'limma'))
install.packages('pheatmap')
library(GSVA)
library(limma)
library(pheatmap)
(2)自建用于GSVA/ssGSEA分析的基因集合。
GSVA包不提供预设基因集,所以我们得自己建房子。本例中使用的原始基因集.gmt文件依旧来自于GSEA官网Molecular Signatures Database。使用的基因集为C2: curated gene sets-Canonical pathways-KEGG subset of CP。
然后进行一番整理,最后成为GSVA需要的list类型的输入基因集:list的每一层为一个基因集,层名为基因集合的名称。(唉还是动用了for循环它老人家,俺仍需努力鸭。
#prepare the database imported from .gmt file.
original_gmt_GSVA <- readLines('c2.cp.kegg.v7.0.entrez.gmt')
strsplit_no_name <- function(gmt.list_layer){
as.character(unlist(strsplit(gmt.list_layer, split = '\t', fixed = T)))[-2]
}
database_list_GSVA <- lapply(original_gmt_GSVA, strsplit_no_name)
for (layers in 1:length(database_list_GSVA)) {
names(database_list_GSVA)[layers] <- database_list_GSVA[layers][[1]][1]
database_list_GSVA[layers][[1]] <- database_list_GSVA[layers][[1]][-1]
}
注:GSVA的基因集要求和GSEA好不一样!看图说话!

(3)基因表达谱的选择和ID转换。
GSVA分析需要输入【expression matrix】而不仅仅是差异基因名/Log2FC数据进行分析。而且其对表达谱的分布有一定要求。
官方文档说明为:【对于RNA-seq,如使用原始整数counts矩阵作为输入,其分布近似泊松,GSVA函数的kcdf参数选择'Poisson';若为log2转换后的FPKM或已经用DESeq2标准化,近似符合高斯分布(正态分布)的数据,则kcdf参数选择'Gaussian'。】
对于芯片数据直接选择'Gaussian'(过时不看)。
关于数据的分布,这里用表达谱中随便选取的一个基因做一下样本间基因表达水平的密度图展示:
图1:原始counts分布(近似Poisson);图2:DESeq2标准化+vst转换(近似Gaussian);图3:仅FPKM标准化(近似Poisson);图4:log2(FPKM+1)(近似Gaussian)。
其实目前认为原始数据的分布更符合负二项而不是泊松分布。Anyway,R包说明文档上说行就行吧,总之是一种近似。
再品味一下上面的参数选择,有感觉了吗~

我们首先选择DESeq2+vst转换表达矩阵进行GSVA:ID转换。用到了GSEA时建立的ENTREZ转换表。
#prepare the normalized counts matrix (rownames are ENTREZIDs of genes)
expr_norm_for_GSVA <- assay(vst(dds_DE, blind = T))
expr_norm_for_GSVA <- data.frame('gene_name'=rownames(expr_norm_for_GSVA), expr_norm_for_GSVA, stringsAsFactors = F)
expr_norm_for_GSVA <- dplyr::inner_join(ENTREZ, expr_norm_for_GSVA, by='gene_name')[,c(-1,-3,-4)]
rownames(expr_norm_for_GSVA) <- expr_norm_for_GSVA$ENTREZID
colnames(expr_norm_for_GSVA) <- gsub(colnames(expr_norm_for_GSVA), pattern = '\\.',replacement='-')
expr_norm_for_GSVA <- expr_norm_for_GSVA[,-1]
(4)GSVA分析。
我们使用GSVA方法来分析(如果想用ssGSEA, method输入'ssGSEA')。GSVA输入的表达矩阵必须是matrix格式,别忘了转换。kcdf参数这里显然就是'Caussian'了。最后一个参数如果电脑好可以设大一点。
#the input data must be in matrix format.
#We should set the distribution as 'Poisson' when using RNA-seq integer counts data rather than the default 'Gaussian'.
#If the matrix is derived from microarray or the RNA-seq integer counts have been transformed to log2 form or..., we can directly set kcdf as 'Gaussian'.
es <- GSVA::gsva(as.matrix(expr_norm_for_GSVA), database_list_GSVA,
mx.diff=FALSE, verbose=FALSE, method='gsva', kcdf='Gaussian',
parallel.sz=1)
(5)limma包筛选差异通路。
limma包,和edgeR,DESeq2并驾齐驱为三大差异分析R包。
limma筛选所需要的东西不多:
(1)es: the pathway score we got from GSVA.
(2)condition_table_for_limma: the matrix containing the grouping info. It contains 2 columns (1 for normal, another for cancer) in this tutorial. Samples (recorded by rownames) affilifated to this group should be 1, otherwise they are tagged with 0.
(3)contrast_matrix: tell limma which the reference group is.
接下来就是线性拟合以及用经验贝叶斯法计算p值。最后我们提取|Log2FC|>0.5,p值<0.05的显著pathways。
#cancer=1, normal=0.
condition_table_for_limma <- model.matrix(~0+ condition_table$sample_type)
colnames(condition_table_for_limma) <- levels(condition_table$sample_type)
rownames(condition_table_for_limma) <- condition_table$TCGA_IDs
#generate the contrast matrix. cancer=1, normal=-1. It means that genes in normal group will be at denominator level.
contrast_matrix <- makeContrasts('cancer-normal',levels = condition_table$sample_type)
lmfit <- lmFit(es, condition_table_for_limma)
lmfit <- contrasts.fit(lmfit, contrast_matrix)
lmfit <- eBayes(lmfit)
#get significant DE pathways.
sigPathways <- topTable(lmfit, coef=T, p.value=0.05, adjust="BH", lfc = 0.5)

(6)热图绘制。
annotation_col <- data.frame('Sample_type'=condition_table$sample_type)
rownames(annotation_col) <- condition_table$TCGA_IDs
pheatmap::pheatmap(t(scale(t(es[rownames(sigPathways),]))), show_colnames = F, show_rownames = T,
annotation_col = annotation_col, cluster_cols = F,
color = colorRampPalette(c("navy", "white", "firebrick3"))(100),
annotation_colors = list(Sample_type=c(cancer = 'orange', normal = 'blue')))
结果说明这组肺鳞癌患者的肿瘤细胞可能存在细胞周期失控、细胞分裂活跃、DNA损伤和修复高度活动的问题。这都是老生常谈的肿瘤特征了,也间接说明这套方法学是非常有效的。

*Optional:不同数据类型进行GSVA分析的比较
根据之前GSVA对数据的要求,我们知道多种数据类型都可以做这套分析。接下来我使用原始counts矩阵,以及z score of log2(FPKM+1)矩阵做GSVA。
#I just show the difference in GSVA analysis. The codes for data processing (like ID conversion, and log2/z_score calculation) have been omitted.
#Integer counts
es_counts <- GSVA::gsva(as.matrix(mRNA_expr_for_DESeq), database_list_GSVA,
mx.diff=FALSE, verbose=FALSE, method='gsva', kcdf='Poisson',
parallel.sz=4)
#z_score(log2(FPKM+1))
es_FPKM_zscore <- GSVA::gsva(mRNA_exprSet_FPKM_log2zscore, database_list_GSVA,
mx.diff=FALSE, verbose=FALSE, method='gsva', kcdf='Gaussian',
parallel.sz=1)
最后得到的差异通路比较如下。虽然略有差异,但是大部分差异通路以及聚类情况都是高度统一的,说明不同数据类型之间的可重复性。大家随意选用。
写了一天...已die...如果觉得好用就点个赞8!