
个体的微生物组在高维空间中随时间变化以形成其个体微生物组云(personal microbiome cloud,Humans differ in their personal microbial cloud)。无论是在平均微生物组概况(一致性)还是云的直径(稳定性)方面,这种云可能与其他人的相似或不相似。然而,目前还没有强大的非参数测试来确定患者与健康个体的微生物组云差异。

在这里,我们提出了在人类肠道微生物组中检测离群值的检验方法,该测试解释了在一组典型的健康个体和个体内时间变异中观察到的广泛的微生物组表型(microbiome phenotypes)。我们强大的非参数离群值检测测试CLOUD测试对患者的微生物组健康状况进行了两次评估:
- 一致性,患者的微生物组云在生态学上与健康受试者的子集相似的程度;
- 稳定性,将患者的云直径与健康受试者的云直径进行比较
测试基于局部线性嵌入式(locally linear embedded)生态距离,使其能够解释参考个体的微生物组云变化。 它还利用患者和参考个体内的时间变异性来增加测试的稳健性。
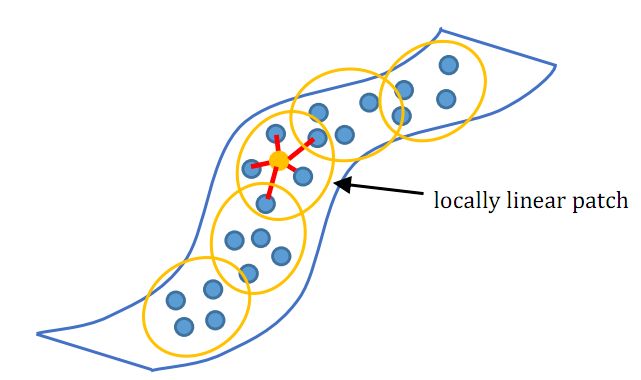
LLE算法认为每一个数据点都可以由其近邻点的线性加权组合构造得到。算法的主要步骤分为三步:(1)寻找每个样本点的个近邻点;(2)由每个样本点的近邻点计算出该样本点的局部重建权值矩阵;(3)由该样本点的局部重建权值矩阵和其近邻点计算出该样本点的输出值。
我们描述了CLOUD测试,并将其应用于一个新的和两个已发表的接受粪便微生物群移植治疗复发性艰难梭菌结肠炎的患者队列,以及两个已知的健康队列,证明了CLOUD 与临床结果具有一致性。尽管CLOUD测试本身并不是临床失调的测试方法,但它仍然为离群值测试提供了一个框架,可用于评估疑似生态失调,这可能在许多儿科和成人疾病的诊断和预后中发挥作用。
实质性方法(Substantial methods )的发展对微生物组的更好的判别测试,其目标是确定区分治疗组或与实验变量或临床元数据相关的特定分类群。当研究具有两个或更多个实验组或与微生物组相关的已知生化梯度时,这些监督测试是有用的。Halfvarson等人最近定义了一个二维健康平面,使用最小二乘法在一个来自健康受试者未加权UniFrac距离的主坐标分析(PCoA)的空间中计算。然后将该平面用作代表健康受试者内正常微生物变异的代表,并总结与间歇性生态失调炎性肠病(IBD)相关的离群值。作者发现,IBD患者的微生物组比健康个体的微生物群波动更多,并且有时根据与新定义的健康平面的偏差占据PCoA空间的不同区域。这种方法代表了生态失调测试的显着进步,并且在具有相对同质和单峰参考群体的情况下可能是有效的。
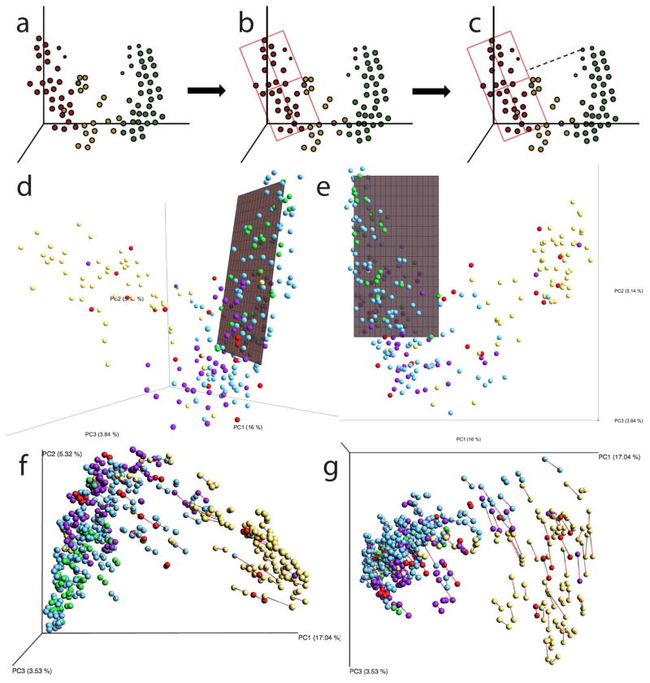
该图总结了基于一组样品S(a,样品选择;b,模型拟合;c,所有样品)的距离计算创建代表平面的过程。
d: 健康平面位于UniFrac空间中将线拟合到点的主轴,
e:定义最小二乘拟合以识别平面,该平面最小化距离最近点的距离的平方和。
f:验证健康平面的位置不是由以蛋白细菌为主的异常值驱动的:Procrustes分析比较原始样本和去除Proteobacteria的样本。 在省略Proteobacteria(黑色)后,载体将每个原始样品(红色)与相同样品连接。 p <0.001,M2 = 0.018,999个排列。
g:大多数载体的短长度表明当过滤掉变形菌时,大多数样品的相对组成不会改变。
实际上,人类微生物组是高度多变量的,并且健康可以与许多不同的分类变量相关联,这些变量可能无法通过平面或超平面捕获。目前还没有已知的微生物组离群值的非参数检验,定义为生态距离空间中的群落状况与健康受试者的大型参考组的显着偏差。这种测试在医学微生物组研究中将是重要的,用于将患者的微生物组与参考群体进行比较,以确定何时在合格性或稳定性方面是显着异常或生态缺陷,而没有对生理状态的先验知识。
在这里,我们提出了基于云的局部线性无偏生态障碍(Cloud-based Locally linear Unbiased Dysbiosis,CLOUD)测试,这是一种利用完整的高维样本间生态距离矩阵的广泛稳健的非参数测试。最终,该测试可以纳入临床实践,以增强基于微生物组的诊断和决策。
方法
CLOUD测试简述
开发用于生态失调的广义测试的一个主要挑战是人类肠道微生物组成在个体之间是高度不同的,一些健康个体具有几乎完全不同的分类群。因此,全群落水平(whole-community level)的生态相似性测量是传统单变量测试(如血液中使用的测量)的合理替代方法。典型的血液检测报告个体血液代谢物的水平,并根据健康个体的正常范围将其分类为正常或异常。

因此,我们的目标是建立一个无监督的多维测试,与健康受试者的参考队列相比,该测试允许将完整的微生物组概况分类为足够健康或离群值。 该测试考虑了以下三个挑战:
- 人类微生物组是多变量的
- 健康的人体肠道微生物组具有许多不同的分类变量(taxonomic configurations)
- 个体的微生物组可以每天大幅变化
我们建议使用非参数CLOUD测试来解决这些问题。具体而言,为了解决第,CLOUD测试使用全群落差异的多变量生态距离测量来代替单个物种的单变量测试。
微生物组的比较是高维度的,而降维分析(例如PCoA)可能完全模糊掉离群值。
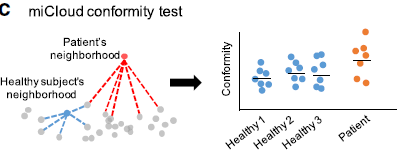
重要的是测量患者微生物组的一致性(与健康个体的相似性)和稳定性(相对于健康个体的概况随时间的一致性的概况随时间的一致性)。与参考微生物组概况相比,测试单个微生物组概况的一致性是一个非常重要的问题。健康个体的微生物组分分布可以占据高维微生物组空间中的任意密度分布。这些分布可以具有曲率,间隙/簇,多个模式和长梯度。因此,符合性的典型参数测量,例如多元正态分布或马哈拉诺比斯距离(multivariate normal distributions or
the Mahalanobis distance),不足以捕获这些复杂的,任意的和高维的密度分布。另一方面,简单的基于质心的测试(其中测试样本与正态分布云的质心进行比较)也可能会根据参考云的形状模糊离群值。
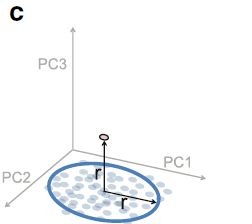
使用生态距离的微生物组离群值的非参数检验
为了解决上述点,测试仅使用生态距离(UniFrac距离或Bray-Curtis距离)来评估测试点与参考云的相似性,而不是点到整个分布距离。 程序如下:
对于大小为n的参考群体中的每个参考对象,在参考群体中识别k个最近邻近的对象。计算邻域的直径,作为从该主体到k个邻居的平均生态距离。 k通常选择为参考集总大小的5%。
计算平均邻域直径
- 对于每个参考对象,计算该对象的邻域直径与平均邻域直径的比率
- 确定参考群体中测试样本的个最近邻居。 计算,从测试对象到其个最近参考邻居的平均生态距离,以及离群检测测试,该对象的邻域直径与参考组中的平均邻域直径的比率:
- 计算测试对象的经验离群值百分位数作为参考离群值检测的分数大于或等于测试对象的离群值检测测试。
换句话说,如果一个人的微生物组与至少少数其他正常人足够接近,并且如果它偏离这种关系就会产生异常,那么它被认为是正常的。例如,离群值百分位数表示测试对象距其最近的个参考邻居更远,而的参考对象来自其最近的个参考邻居。 离群值检测统计量也具有简单且有用的解释: 具有离群值检测测试的对象的邻域直径是参考群体中的平均邻域直径的两倍。
测试的一个重要特征是它仅利用生态距离空间中的局部距离。这使得它能够非参数地考虑健康微生物组的高维度的任意密度分布.较大的值通常与推定的离群值的数量增加相关,即使在参考分布中也是如此。 对于下面描述的这个和其他测试,将设置为接近完整数据集的大小允许从临床角度对离群值进行最保守的识别。然而,远小于受试者总数的值允许测试考虑正常微生物组概况中更大的全局变化。因此,可以被认为是高维参考微生物组云的形状上的平滑参数。通常,应至少大于参考分布中预期的离群值的数量。在我们的标准测试中,我们在测试单个样本时将设置为参考样本总数的5%,并且在平均每个主题内的样本之间的距离时将参考主体总数的5%.我们还在下面描述的几个数据集上测试了几个值,对应于群组的5-80%的范围,并且发现结果对的选择不是特别敏感。
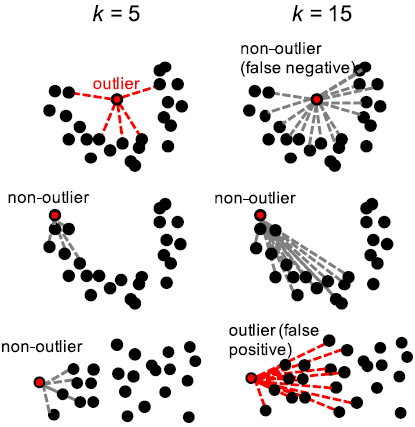
某些高维数据设置太高的图形说明可以导致实际离群值被归类为正常(假阴性),并且可以导致正常点被分类为离群值(假阳性)。
使用大逼近的目的是破坏局部距离度量,这是为了允许测试仅使用生态距离空间中的局部区域,并且可以将分布的极值处的正常参考样本归类为离群值。
另一方面,如果太小,那么它对参考组中的细微变化不具有鲁棒性。默认情况下,云测试将邻域大小设置为参考集大小的5%。
使用的目的是测试的关键组成部分,因为它允许测试的灵活性相对于参考群体的生态距离所在的高维流形中的任意形状.较大的样本量对于增加功效始终很重要。 更大样本量有利于测试的方式是参考群体的大小。 整个参考人群可用于测试任何单个测试对象以确定它是否是离群值,但使用局部邻域对于避免与基于质心的测试相关的陷阱至关重要.
为了解决上述点,我们还在此处介绍了稳定性测试.与一致性测试一样,对来自一组测试受试者的每个测试受试者分别进行稳定性测试。我们使用自相似性,通过测量受试者在该受试者前一天的1天的生态距离(例如,未加权的UniFrac距离)来计算日常稳定性。然后将测试对象的所有日常UNIFRAC自距离的平均值与参考对象的平均每日UniFrac自距离的分布进行比较,以获得与一致性测试相同的经验离群值百分位数。
患者于与志愿者
我们分析了几个公布的数据集以及来自参考群体的新样本。这些患者包括来自一个公布的数据集的五名患者,他们患有多重难治性梭状芽孢杆菌感染(CDI)难治于标准抗生素治疗(患者CD1至CD5),并用粪便微生物移植治疗。在接受CDFI的五例患者中,四例FMT治愈,1例失败。十六名健康受试者参加明尼苏达大学微生物群标准献血员。治疗方案也参与了这项研究。先前描述了粪便供体资格的排除和排除标准。简而言之,除了作为献血者的资格之外,这些人不服用药物;无近期(6个月)抗生素暴露史;无胃肠道,无免疫力,神经发育或精神问题;体重指数<25 kg/m2;代谢试验正常。明尼苏达大学机构审查委员会(IRB)批准了粪便样本的前瞻性收集和分析。
粪便微生物区系移植(Fecal microbiota transplantation,FMT)
FMT是通过标准化制备浓缩新鲜或冷冻粪便细菌,如先前描述的结肠镜检查进行的。所有患者均口服口服万古霉素,每日125次,每次四mg,直至术前2天。
在手术前一天,患者接受聚乙二醇结肠镜检查(GoyType®或MOVIPREP®),以去除残留抗生素和粪便材料. 通过结肠镜活检通道将供体粪便微体置于回肠末端和或盲肠。
样本选择
使用拭子收集粪便样品,以在生产后立即获得粪便沉积到厕帽中。随后将样品转移到实验室,按照先前描述的方法进行处理,并在80℃储存直到使用。从FMT手术治愈的4例复发性CDI患者中,从第2天(前2天)到第151天(151个FMT)共收集96个样本。此外,收集了59例FMT未能治愈的患者FMT后的FMT标本。我们还收集了247例健康受试者的粪便样品,从第1天(收集的第一天)到第75天。
DNA提取、PCR、测序及序列处理与分析
- 16S rRNA V4 region
- 97% similarity cut-off against Greengenes
- analyzed by using unweighted UniFrac, followed by PCoA
- R version 3.4.0
结果
解释测试
测试提供零假设的离群值百分位数,即单个预定测试对象的微生物组概况来自独立参考群体。离群值百分位数描述了随机选择的健康受试者具有与测试对象一样大的邻域大小的概率。离群值百分位数由参考人口中邻域大小的经验分布确定。参考群体中包括的参考对象组由重复随机采样获得。
可以考虑类比,根据人们参考人群中观察到身高的分布,将离群值百分位数分配给一个人的身高。
如果参考群体中的人的高度是正态分布的,则可以使用正态分布来为测试对象分配离群值百分位数。在假设参考对象身高遵循具有某些参数的正态分布的情况下,该离群值百分位数将描述参考对象的哪个部分具有大于或等于测试对象身高。如果这种正态性假设对于特定的参考群体是错误的,并且如果参考群体足够大以获得小的离群值百分位数并且足够无偏见地代表总参考群体的真实随机抽样,那么可以改为使用经验参考组中身高的分布,以获得独立测试对象的经验离群值百分位数。
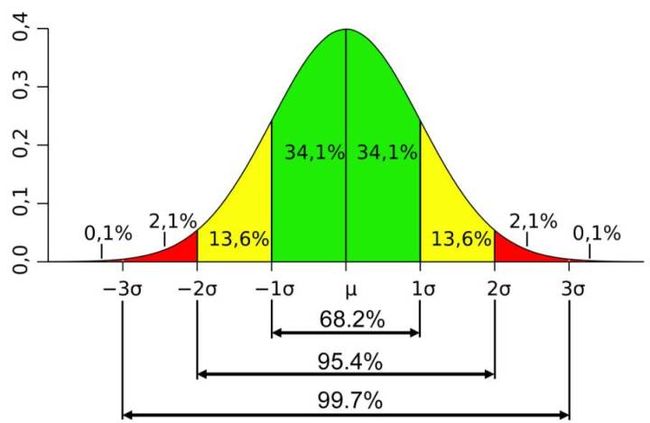
以相同的方式,CLOUD测试离群值百分位数仅仅是参考对象的分数,其局部邻域直径大于或等于独立测试对象的邻域直径。
重要的是,我们的测试不是用于识别参考人群中的离群值,尽管我们确实执行了保持交叉验证来评估由来自三个不同国家的人组成的健康人群中的异常状态,以证明的灵活性测试参考组中的聚类和多变量变化。
有很多已建立的统计方法,旨在识别给定参考组内的离群值。
这些包括用于测试是否存在单个离群值的Grubbs测试(Grubbs' test for outliers)
,用于测试是否存在特定数量的离群值的Tietjen-Moore测试(TIETJEN-MOORE TEST
),以及用于测试是否存在任何数字的广义极端学生化偏差测试(t.test),低于某个上限在一组正常分布的参考值中存在的离群值。
与这些测试相反,测试假定参考集没有离群值,而是设计用于测试单个新的独立主题是否是基于参考集的离群值。
与前述建立的测试相比,测试是多元的,非参数的,没有关于参考值分布的假设,并且基于专门设计用于比较群落组成的生态距离度量。
生态距离矩阵相对于分类轮廓的维数
与使用少量主坐标分析(PCoA)空间维度的离群值检测方法相比,测试确实使用了生态距离矩阵的全部数据,没有任何降维操作。
距离度量本身是数据从P维空间的变换,其中P是微生物组概况中的分类群的数量,到“N-1”维空间,其中N是样本的数量。
取决于参考群体的大小,P有时可能基本上大于N,并且距离变换将表示将分类法简档嵌入到较低维度空间中。
例如,如果只有100个样本的数据集中有1000个类群,那么生态距离矩阵可能具有比分类子轮廓矩阵更低的秩;
然而,在类群分布矩阵中经常有许多类群的相关组,使得分类单元轮廓矩阵的实际秩可能小于所观察到的唯一分类群的数量。
因此,使用完整生态距离矩阵的测试不一定利用分类单元轮廓空间的全维度,但确实利用比仅在少量PCoA维度中操作的测试大得多的维度。
应用1:健康受试者的一致性测试
为了评估我们的测试鉴定健康个体的能力,鉴于参考人群差异很大,我们使用两个大规模的微生物组数据集来填充健康微生物组的多维景观。
然后,我们使用保持测试( [hold-out testing](https://zhuanlan.zhihu.com/p/37646822)来评估测试的I型错误率,将这些参考群体重复子采样到单独的“参考”和“测试”组中。
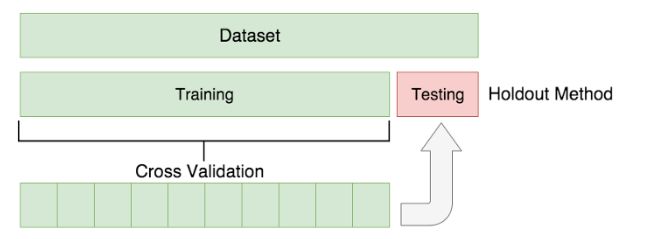
什么是保持测试 ? 从100个例子里硬生生抽出20个例子把他们晾在一边,完完全全不参与模型训练。这20个例子我们称之为hold-out set。然后剩下80个例子,等分成4份然后做4次模型训练。每次都用到不同的25% 的测试集(剩下的75%则作为训练集)。 【该方法也称作k-fold cross validation,本例中,k=4】。最终,在四次训练的基础上,优化参数,取平均值。等模型训练好后,用这个hold-out set来证明给别人看:我的模型是可以很好地工作在没有参与模型训练的例子上滴!
首先,我们分析了人类微生物组项目(HMP)的16S rRNA基因数据(可变区V3-V5),包括239名健康受试者
这些数据可在https://www.hmpdacc.org/上获得。 在该数据集中,我们使用肠道样本的子集,不包括来自肥胖患者的样本,从200个患者中留下200个样本。 HMP DACC网站(https://www.hmpdacc.org/HMMCP/)上提供了完整的元数据和注释协议。 我们使用200个粪便样本的未加权UniFrac距离矩阵作为生态距离矩阵。
虽然CLOUD测试旨在将测试对象与独立参考组进行比较,但我们希望评估参考人群子集相对于其余参考人群的离群值状态。
为了达到这个结果,我们随机抽取50名受试者作为测试用例,然后对其他150名受试者进行二次抽样,直至100次训练,并重复该过程30次。
在30个重复的过程中,使用这些随机选择的训练集,我们将CLOUD符合性测试应用于k的几个值(最近邻居的数量),从到(所有测试群组-1)并且没有识别任何 受试者作为离群值,除了几个随机数据集中的k的极值,证明了CLOUD检验对邻域大小的稳健性和低假阳性率
我们还评估了以前发表的马拉维农村和美国大都市地区的个人测试。
我们仅包括来自15岁以上受试者的粪便样本(n = 219)。 我们使用了219个粪便样本的未加权UniFrac距离矩阵。 我们随机抽取50名受试者作为测试用例,然后对其他169名受试者进行二次抽样,直至100次训练,并重复该过程30次。
在30个重复的程序中,使用随机选择的训练数据集,我们将生态失调测试应用于k的若干值,如上所述,并且在任何训练/测试子集中没有发现离群值。 这证明了测试对来自给定参考群体的不同训练集的稳健性。 在这里,该测试可以成功地解释非常高的个体间差异,因为来自不同国家的受试者具有高度不同的微生物组。
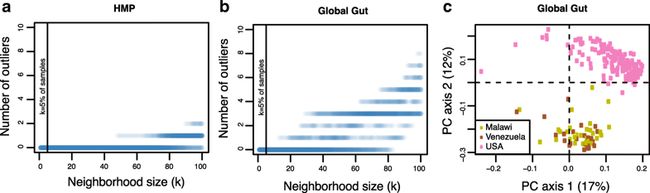
a在测试预测中选择的最近的健康邻居数,以查找国际群组中的离群值(HMP数据集)。 我们随机选择50名受试者的测试数据集,并在200名受试者的完整数据集中随机选择100名受试者的训练数据集30次。 我们重复训练数据集的随机选择30次。 除了几个随机训练数据集中的k的极值之外,我们没有识别离群值。 该分析证明了CLOUD测试对邻域大小的稳健性。 垂直条表示训练数据集的5%,即测试的默认邻域大小。
b对全球肠道数据集进行了相同的分析。
c来自Unweighted UniFrac距离的全局肠道数据集的主坐标图,证明CLOUD测试对于具有参考组的强聚类效果是稳健的
应用2:FMT后的微生物组修复
在人类和小鼠模型中,粪便微生物群移植(FMT)已经证明了治愈CDI的高效率,CDI是一种严重且复发的感染,发病率增加。 一些研究报道,FMT后接受者的粪便微生物组比移植前收集的患者的微生物组更加多样化并且与供体微生物群落结构更相似。 我们小组最近的一项研究表明,FMT导致细菌粪便样品组成从明显的生理状态快速正常化,成为正常粪便微生物组的一个代表,成功用FMT治疗。 然而,没有良好的统计学测试来确定患者的微生物组相对于健康受试者群体是否已经恢复。 在这里,我们将测试应用于FMT接受者,并将其与一组健康受试者的一致性进行比较。
一致性和成功的FMT
我们应用CLOUD测试来评估我们队列患者中FMT后成功的微生物组恢复。 下图中的结果绘制了10个最近的独立健康邻居,显示在FMT后治愈CDI的患者中非生理性微生物组的明显恢复,因为两组中未加权UniFrac距离非常接近(健康受试者) 和成功的FMT)和平均距离之间的差异没有什么不同。 这也显示FMT在最终复发CDI的患者中恢复微生物组失败,因为这两组之间的距离明显不同(健康受试者和FMT失败,离群值百分位数<0.001)。 这表明CLOUD测试能够成功区分FMT最终导致成功的患者。
然后,我们在每个患者的水平上应用测试,即将来自单个患者的所有样本聚合成单个平均样本(FMT的响应者和非响应者),并且发现四个响应者患者不被视为离群值, 而对FMT无反应的患者被认为是离群值。 这种一致性测试对于邻域大小是稳健的,因为增加最近的独立健康邻居的数量(从到)总是显示健康对照与来自未成功用FMT治疗的患者的样本之间的显着差异.且健康对照和响应FMT的患者的样品之间没有区别。 然而,使用非常大的,一名应答者患者被认为是离群值(离群值百分位数<0.05)。 同样,这说明了只使用生态距离的局部邻域是可行的。
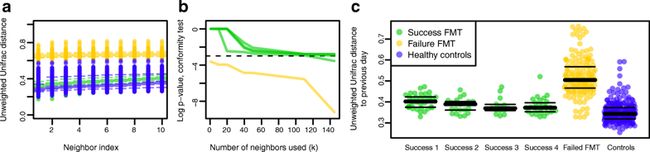
a前10个最近的独立健康邻居。该图显示了FMT响应者中微生物组的恢复,因为健康受试者和FMT成功的样本之间的距离非常相似。这也显示无应答患者中微生物组的恢复失败,因为来自失败患者和健康受试者的样品之间的距离非常不同。
b接受FMT的患者log10离群值百分位图。虚线表示0.05的离群值百分位数。当使用k = 5%的人群时,无应答者患者被认为是离群值。使用大的邻域大小将1个响应者患者分类为离群值。
c通过随时间的自相似性测量的患者稳定性。使用Unweighted UniFrac距离绘制一天到相应前一天的距离。该图显示健康对照和响应者患者中两个连续粪便微生物组样本之间的稳定性,而无应答者患者在两个连续样本之间显示不稳定
在其他两项FMT复发CDI研究一致性检验
我们将测试应用于描述复发性CDI的已发表数据集,该数据集探讨了FMT粪便供体和受体的粪便微生物群。该数据集包括来自供体的10个样品,来自接受者的14个FMT前样品和来自FMT后样品的16个样品。具体而言,5个FMT后样本检测为伴随艰难梭菌(Clostridium difficile)阳性,11个FMT后样本检测为阴性。我们使用捐赠者队列来定义最近的独立健康邻居。使用Bray-Curtis距离,我们测试了从FMT接收者收集的所有样本。我们发现,当k对应于健康供体样品数量的5%至40%时,检测为伴随艰难梭菌的阳性的FMT后样品均被CLOUD视为离群值(离群值百分位数<0.001)。此外,FMT前样本被认为是离群值(离群值百分位数<0.001),而同时艰难梭菌(离群值百分位数= 0.4至0.75)检测为阴性的FMT后样本被正确归类为非离群值。
我们还将测试应用于另一个已发表的粪便数据集,该数据集描述了来自肠道微生物组的预测信号与复发性CDI的发展之间的关系。该数据集包括来自供体的10个样品,从CDI复发的患者收集的11个受体样品,以及来自未发现复发的患者的21个受体样品,因为它们被认为是非遗传性和治愈的。我们使用来自相同数据集的捐赠者群组来定义最近的独立健康邻居。使用粪便样本的Bray-Curtis距离矩阵,我们测试了在FMT接收者中收集的所有样本。与之前的数据集一样,我们发现,当k相当于健康供体样本数量的5%到40%时,来自复发患者的样本被CLOUD视为离群值(离群值百分位数<0.001),所有前FMT受体样本(离群值百分位数<0.001)。相反,来自未复发的患者的所有样本均符合(不考虑离群值,离群值百分位数= 0.6至0.65)。
FMT数据集中的稳定性测试
为了评估通过FMT治愈CDI的患者肠道微生物组的稳定性,我们获得了上述稳定性度量。 我们观察到健康对照中粪便微生物组在每日时间过程中以及在FMT手术后几天内成功应答FMT的患者中的高稳定性。 相比之下,对FMT没有反应的患者的微生物组在FMT手术后的不同样本时间集合中显示出生态失调和不稳定性。 上图c中的结果显示健康对照和成功应答FMT的患者的粪便微生物组的连续每日样品之间的高稳定性,而复发的患者在每两个连续样品之间平均显示出显着更高的不稳定性,尽管存在不足 具有多个每日时间点的独立参考受试者的数量以获得可靠的离群值百分位数。
讨论
随着时间的推移,个体之间和个体之间的微生物组组成存在很大差异。已经开发了用于测试疾病状况是否与特定分类群或整体生态群落组成相关或相关的方法。然而,据我们所知,基于健康个体的参考组,在给定时刻或在给定时期内微生物组谱的变化很大,以前没有发表非参数统计检验,表明患者的整体微生物组概况是否可以被认为是健康的。
在这里,我们通过表征人肠道微生物组来开发一种从FMT后的生态状态恢复的测试,该微生物组解释了在一组健康个体中观察到的广泛的微生物组表型以及个体内的时间变化。这种稳健的非参数测试基于当地生态距离,可用于识别具有微生物组的受试者,所述微生物组在一致性或随时间稳定性方面显着异常。我们的测试进一步允许无监督检测微生物组离群值。我们已在三个临床数据集中验证了生态失调试验,以显示生态失调试验与艰难梭菌感染复发的临床结果的一致性。我们还证明,随着时间的推移,受试者的微生物组云内的局部稳定性分析提供了经历成功和不成功的FMT程序的患者的强烈分离,其中失败的程序(定义为晚期CDI复发的存在)导致患者微生物组明显不太稳定。
由于三个原因,这种无监督的微生物组分析中的符合性和稳定性离群值的识别特别具有挑战性,我们已经在我们的方法中解决了如下问题。
首先,人类微生物群是高度多元的,每个个体中包含成百上千种不同的物种。我们的生物障碍测试使用生态学和系统发育的完整微生物群落距离度量,如UNIFRAC(用于操作分类单位或OTU)或Bray Curtis(物种级分类),以评估两个个体内的物种或OTU混合物的散度水平,而不是关注微生物群中的任何个体成员。
第二,健康的人类肠道微生物群有许多不同的分类学结构。两个人可以有几乎完全不同的细菌集合,但仍然可以被认为是健康的。我们的方法只使用生态距离。我们评估测试对象的生态邻近度与该对象最近的健康邻居,以确定测试对象的“个人微生物群落云”的一致性。然后,我们比较云的接近所有健康的人的云,以确定测试对象是否足够接近至少其他一些健康的人,以被认为是健康的。只依赖于局部生态距离允许灵活性来解释任意尺寸和密度分布的高维集的个人微粒体云团的健康个体。
第三,个体的微生物组可以每天大幅变化。我们计算上面的邻域大小不是基于来自每个主题的单个时间点,而是基于多个时间点的平均值来计算主题的微生物组云一致性的时间变化。此外,我们建议对受试者个体微生物组云的直径进行单独测试,并将其与参考或健康受试者的微生物组云的直径分布进行比较,以评估稳定性。
第四,在研究中难以以完全相同的方式收集和储存所有样品,特别是在纵向研究中,在最终时间点收集的样品在DNA提取之前在冷冻储存中花费的时间少于在其他时间收集的样品。在小鼠研究中,笼养和动物批次效应也会引入系统性偏差。 测试可能是一种有用的方法,用于检测研究中的离群值,其中有问题的数据与样本收集或保存错误相关。
测试有几个值得注意的限制。 该方法的关键组成部分是所使用的距离度量的选择,因为不同的距离假定了不同的生态相似性模型。 在这里,我们使用Unweighted UniFrac距离度量,因为我们分析16S数据,UniFrac距离是这种情况下的有效距离度量。 然而,其他生态距离可能适合某些研究,事实上,我们发现Bray-Curtis在CLOUD测试中很好地区分了两个复发CDI数据集中的恢复和非恢复。 CLOUD测试还要求正确选择参考集以表示高维参考微生物组景观的充分变化,并且以与参考样品相同的方式收集和分析测试样品。
结论
随着医学微生物组研究领域越来越接近从流行病学调查到临床应用的转化,临床医生需要一种可靠的测量方法,可以确定微生物组是否与参考人群中的微生物组在统计学上相似。该测量必须考虑微生物组的高维度,高个体间可变性和高纵向可变性。 CLOUD测试旨在解释这些限制因素,可用于将患者的微生物组与参考人群进行比较,以确定其在一致性或稳定性方面是否显着异常或是生理异常。该测试依赖于具有健康个体的相关参考群组,但由于其依赖于本地距离而对于从数据库中添加或去除高度不一致的样本也是完全不变的。检测与整合或稳定性相关的生态失调的能力可以作为在儿科或成人临床实践中与微生物组功能改变相关的各种医学病症中的诊断工具。
文章中大牛给出了计算离群值百分位数的R函数,只要输入一个距离矩阵就可以啦:
# inputs a distance matrix
# returns piecewise distances of samples and their outlier percentile
# and a matrix of the repeated measures of distances
# k is number of neighbors choosen
"piecewise_kn_V1" <- function(d, test.ix, k=X, ndim=-1){
if(class(d) != 'matrix') d <- as.matrix(d)
stats <- numeric(length(test.ix))
pvals <- numeric(length(test.ix))
for(i in 1:length(test.ix)){
ref.ix <- test.ix[-i]
keep.ix <- c(test.ix[i], ref.ix)
if(ndim > -1){
pc <- cmdscale(d[keep.ix,keep.ix,drop=F],k=ndim)
d.i <- as.matrix(dist(pc))
} else {
d.i <- d[keep.ix,keep.ix,drop=F]
}
test.dist <- mean(sort(d.i[1,-1])[1:k])
ref.dists <- numeric(length(ref.ix))
for(j in 1:length(ref.ix)){
ref.dists[j] <- mean(sort(d.i[-1,-1][j,-j]))
}
stats[i] <- test.dist / mean(ref.dists)
pvals[i] <- mean(test.dist < ref.dists)
}
result <- list()
result$stats <- stats
result$pvals <- pvals
outcome <- pvals <= 0.05
result$lenght <- length(outcome[outcome==TRUE])
return(result)
}
CLOUD: a non-parametric detection test for microbiome outliers
机器学习降维算法三:LLE (Locally Linear Embedding) 局部线性嵌入
Emmanuel Montassier
Dan Knights
Grubbs' test for outliers
一个Outlier的江湖 -- 经典统计观
离群值!离群值?离群值!
分辨真假数据科学家的20个问题及回答
TIETJEN-MOORE TEST
机器学习中testing和hold-out的区别【为什么要分出一个hold-out】
103.3.4 Box Plots and Outlier Detection
Outliers: Finding Them in Data, Formula, Examples. Easy Steps
