最近在做某个植物小物种的miRNA分析,看过几个公司的miRNA分析报告和文献后开始动手。大概分以下几步:
1、筛选出目标miRNA
(a)根据长度筛选clean data(植物18~30nt)
(b)比对到参考基因组上,去除未匹配序列
(c)比对到Rfam中的ncRNA,去除snRNA,snoRNA,rRNA,tRNA等
(d)去除重复序列
2、保守miRNA的鉴定(与mirbase数据库比对)
3、新miRNA的预测
4、差异表达(和mRNA流程类似)
5、靶基因的预测(植物用psRNATarget在线分析)
这其中第4、5步我比较熟悉,前三步稍微陌生一点。能找到软件整合流程一步跑下来最好,不行的话只能分步尝试(第一步如果参考基因组注释比较完整,还可以将sRNA比对到mRNA的外显子和内含子,找出来自mRNA降解片段的sRNA并去掉)。
还好我找到了很好用的miR-PREFeR,分享在这里~·github主页https://github.com/hangelwen/miR-PREFeR
miR-PREFeR: microRNA PREdiction From small RNAseq data
miR-PREFeR成功解决了植物miRNA分析中的几大痛点:1. 假阳性高; 2. 耗时长; 3. work only for genomes in their databases; 4. 难用 (亲身经历证明确实好用)。该软件于14年发表在Bioinformatics上并仍在改进中 http://bioinformatics.oxfordjournals.org/content/30/19/2837.abstract.
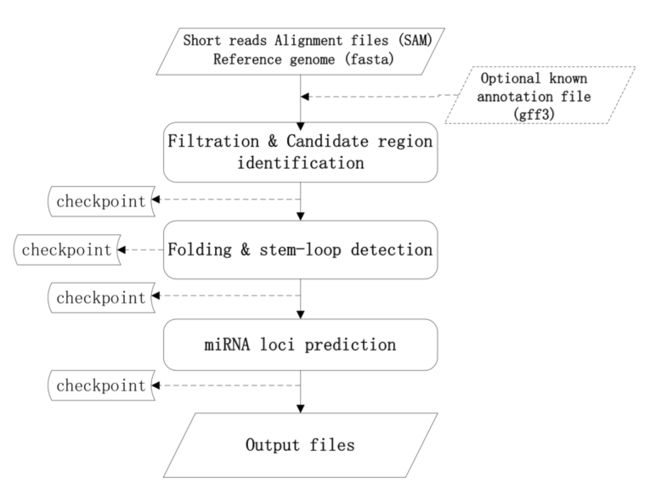
1、前期准备
安装samtools
git clone git://github.com/samtools/samtools.git
安装ViennaRNA ( 最好是1.8.5或2.1.2, 2.1.5版本)
#我装的是最新版
wget https://www.tbi.univie.ac.at/RNA/download/sourcecode/2_4_x/ViennaRNA-2.4.10.tar.gz
tar zvxf ViennaRNA-2.4.10.tar.gz
cd ViennaRNA-2.4.10
./configure --prefix="/user/tools/ViennaRNA/" --without-perl
make
make install
2、安装
git clone https://github.com/hangelwen/miR-PREFeR.git
安装很方便,可以在example里测试一下pipeline
3、输入数据处理
一共需要的输入数据有三个:
参考基因组fasta文件
small RNAseq 数据比对基因组的sam文件(bowtie)
gff文件(可选,记录需要被屏蔽掉的信息,比如重复序列)
其中在比对前,需要把fasta处理一下,samplename.txt里面记录取样信息:
$ vi samplename.txt
root #SAMPLE1.fasta
flower #SAMPLE2.fasta
salt #SAMPLE3.fasta
$ python scripts/process-reads-fasta.py samplename.txt SAMPLE1.fasta SAMPLE2.fasta SAMPLE3.fasta
得到 SAMPLE1.fasta.processed文件,>开头包含sample name、sequential number和depth信息
$ less SAMPLE1.fasta.processed
>root_r0_x1 #sample name_sequential number_depth
ACTACTGCAAGGGCTGGCTCAACCCGC
>root_r1_x5
TGGTTGCTGTCGCTGGTCGCTGGT
4、bowtie比对
python bowtie-align-reads.py -f -r TAIR10.fas -t bowtie-index/ -p 8 SAMPLE1.fasta.processed SAMPLE2.fasta.processed SAMPLE3.fasta.processed
生成pdep.chr1.sam,pind.chr1.sam,cold.chr1.sam
5、准备configuration文件
每个参数都有详细说明
$more config.example
#example configuration file for the miR-PREFeR pipeline.
#lines start with '#' are comments
#miR-PREFeR path, please change to your path to the script folder.
#Absolute path perfered.
PIPELINE_PATH = /sRNA/miR-PREFeR/
#Genomic sequence file in fasta format. Absolute path perfered. If a path relative if used, it's relatvie to the working directory where you execute the pipeline.
FASTA_FILE = ./TAIR10.chr1.fa
#Small RNA read alignment file in SAM format. The SAM file should contain the SAM header. If N samples are used, then N file names are listed here, separated by comma. please note that before doing alignment, process the reads fasta files using the provided script 'process-reads-fasta.py' to collapse and rename the reads. Absolute path perfered. If a path relative if used, it's relatvie to the working directory where you execute the pipeline.
ALIGNMENT_FILE = ./cold.chr1.sam, ./pdep.chr1.sam, ./pind.chr1.sam
#GFF file which list all existing annotations on genomic sequences FASTA_FILE. If no GFF file is availble, comment this line out or leave the value blank. Absolute path perfered. If a path relative if used, it's relatvie to the working directory where you execute the pipeline.
#CAUTION: please only list the CDS regions, not the entire miRNA region, because miRNAs could be in introns. This option is mutual exclusive with 'GFF_FILE_INCLUDE' option. If you have a GFF file that contains regions in which you want to predict whether they include miRNAs, please use the 'GFF_FILE_INCLUDE' option instead.
GFF_FILE_EXCLUDE = ./TAIR10.chr1.CDS.gff
# Only predict miRNAs from the regions given in the GFF file. This option is mutual exclusive with 'GFF_FILE_EXCLUDE'. Thus, only one of them can be used.
#GFF_FILE_INCLUDE = ./TAIR10.chr1.candidate.gff
#The max length of a miRNA precursor. The range is from 60 to 3000. The default is 300.
PRECURSOR_LEN = 300
#The first step of the pipeline is to identify candidate regions of the miRNA loci. If READS_DEPTH_CUTOFF = N, then bases that the mapped depth is smaller than N is not considered. The value should >=2.
READS_DEPTH_CUTOFF = 20
#Number of processes for this computation. Using more processes speeds up the computation, especially if you have a multi-core processor. If you have N cores avalible for the computation, it's better to set this value in the range of N to 2*N.
#If comment out or leave blank, 1 is used.
NUM_OF_CORE = 4
#Outputfolder. If not specified, use the current working directory. Please make sure that you have enough disk space for the folder, otherwise the pipeline may fail.
OUTFOLDER = example-result
#Absolute path of the folder that contains intermidate/temperary files during the run of the pipeline. If not specified, miR-PREFeR uses a folder with suffix "_tmp" under OUTFOLDER by default. Please make sure that you have enough disk space for the folder, otherwise the pipeline may fail.
TMPFOLDER = /tmp/exmaple
#prefix for naming the output files. For portability, please DO NOT contain any spaces and special characters. The prefered includes 'A-Z', 'a-z', '0-9', and underscore '_'.
NAME_PREFIX = TAIR10-example
#Maximum gap length between two contigs to form a candidate region.
MAX_GAP = 100
# Minimum and maximum length of the mature sequence. Default values are 18 and 24.
MIN_MATURE_LEN = 18
MAX_MATURE_LEN = 24
# If this is 'Y', then the criteria that requries the star sequence must be expressed is loosed if the expression pattern is good enough (.e.g. the majority of the reads mapped to the region are mapped to the mature position.). There are lots of miRNAs which do not have star sequence expression. The default value is Y.
ALLOW_NO_STAR_EXPRESSION = Y
# In most cases, the mature star duplex has 2nt 3' overhangs. If this is set to 'Y', then 3nt overhangs are allowed. Default is 'N'.
ALLOW_3NT_OVERHANG = N
#The pipeline makes a checkpoint after each major step. In addition, because the folding stage is the most time consuming stage, it makes a checkpiont for each folding process after folding every CHECKPOINT_SIZE sequences. If the pipeline is killed for some reason in the middle of folding, it can be restarted using
#'recover' command from where it was stopped. The default value is 3000. On my system this means making a checkpoint about every 5 minutes.
CHECKPOINT_SIZE = 3000
6、跑流程
python miR_PREFeR.py -L -k pipeline configfile
pipeline里包含prepare, candidate, fold, predict四步。如果某步中断了,还可以续跑
python miR_PREFeR.py -L recover configfile
7、输出结果
result文件夹里包含如下文件:


吐槽几个大坑:
mirdeep2整合了bowtie、ViennaRNA、randfold、PDF-API2等多个软件,动物miRNA分析用的多,有专门为植物开发的mirdeep-P,但是目前下载不了。而且mirdeep2的安装很死板,路径受限各种报错不好装,本来都装好跑通了后来重新跑的时候就又要重新装一遍,不推荐。
新miRNA预测软件mireap核心脚本使perl写的,里面的模块FFW1现在找不到了,所以用不了
参考:
一个简易的miRNA分析流程