一、格式介绍
(一)gtf 文件。GTF 为General Transfer Format缩写,跟 GFF2格式类似。相信大家做转录组分析时候经常会看到Cufflinks或者Stringtie软件对转录组进行定量与组装会时产生一个gtf文件,里面包含的信息如下:
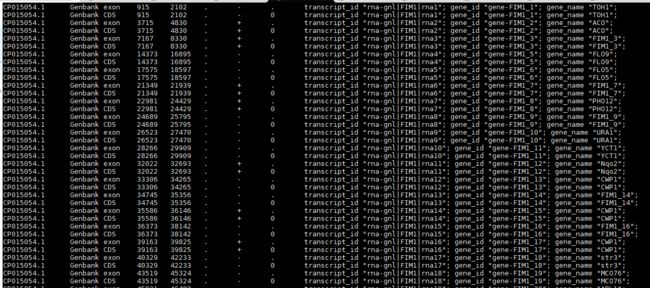
每列信息的含义如下:
seqname - 序列的ID,可以是染色体的ID也可以是Scaffold或者Contig的ID。
source - 产生此文件的软件,如Stringtie产生的则为Stringtie,CUfflinks产生的则为Cufflinks,不知道的使用点 “.” 表示。
feature - 后面start和end之间区域代表的特征,如果此区域是基因,则此处为gene,如果是外显子,则为exon,如果是转录本,则为transcript,如果是非编码RNA则为lncRNA,如果是重复序列,则为TE,等等,主要表明这一块区域的特征。
start -上述feature的在序列上的起始位置。
end - 上述feature的在序列上的终止位置。
score - 一个浮点数值,也可以为点 “.” 。有值的时候代表上述feature的可靠
性。因为无论是gene还是mRNA,都是基于预测差生的,因而必然会有一个值来衡量预测准确性。
strand - + (forward)或者 - (reverse),代表上述feature是位于正链还是负链上。
frame - 内含子相位,只能为'0', '1' or '2',或者为点 “.”。 '0' 代表feature起始碱基为三联体密码子的第一个碱基, '1' 代表三联体密码子的第2个碱基, 2代表第3个碱基。
attribute -备注列。主要备注该feature的一些信息,常见的是gene或者transcript等的ID信息以及FPKM值等,多个备注信息之间通常用分号分隔。
(二)gff 格式。为general feature format缩写,目前采用的是version 3,即我们常说的gff3文件。该文件常用来对基因组进行注释,表示基因,外显子,CDS,UTR等在基因组上的位置。众多基因预测软件如Glean,EVM,AUGUSTUS等会产生此格式文件。
与gtf文件不同之处只是在第9列。此列格式为“标签=值”(tag=value),标签与值之间用“=”,不同的标签之间用“;”隔开,一个标签可以有多个值,不同值用“,”分割。
二、gtf与gff转换以及对GFF文件进行过滤。
常采用的软件是gffread,为Cufflinks自带的一个程序,他不仅可以实现GTF与GFF的互相转换,而且还可以对GFF文件进行过滤处理。以下是gffread的帮助信息:
Usage:
gffread
[-o
[-CTVNJMKQAFGUBHZWTOLE] [-w
[-i
常用参数介绍:
-g 序列文件,即GFF/GTF文件第一列ID对应的序列文件。
-i 丢弃掉内含子大于的转录本(mRNA/transcript)
-r 起始和终止位置,填写示例100.10000即为输出与100到10000有重叠的所有转录组,也可以限制序列ID及链,填写示例:+Chr1:100..10000。
-R 丢弃掉此范围的转录本,与-r相反。
-U 丢弃掉 single-exon的转录本
-C 丢低调无CDS的转录本。
-V 丢弃掉含有移码突变的转录本。
-H 如果使用了-V,则重新检查并调整内含子相位,避免由于翻译起始位点选择的位置不对导致移码突变的产生。
-B 如果使用了-V, 对于单外显子基因,则重新检查相反的链,是否存在移码突变。
-N 丢弃掉多外显子基因剪接位点不是常见的 GT-AG, GC-AG or AT-AC序列。
-J 丢弃掉没有起始密码子或者终止密码子的转录本,仅保留有完整编码框的转录本。
--no-pseudo:过滤掉含有 'pseudo' 的注释信息
-M/--merge : 合并完全相同的或者存在包含关系的转录本。
-d:使用 -M ,将合并信息输出到文件中
--cluster-only: 类似于 --merge 但是不合并转录本
-K 对于-M 选项:also collapse shorter, fully contained transcripts
with fewer introns than the container
-Q 对于-M 选项:移除包含关系的转录本的限制条件:多外显子转录本将会合并,如果他们内含子位置完全一样,单外显子转录本只需要有80%一样即可合并。
--force-exons: 使GFF features的最小层级为exon
-E 对于重复的 ID或者 GFF/GTF 其他潜在的格式问题给出警告信息。
-Z 将内含子小于4 bp的邻近的两个外显子合并为一个。
-w 输出每个转录本的外显子序列
-x 输出CDS序列
-W 对于 -w 和 -x 选项,输出外显子位置坐标到FASTA序列的ID中
-y 输出蛋白质序列
-L 将Ensembl GTF 转换为 GFF3 conversion (implies -F; should be used with -m)
-o 输出"filtered" 后的GFF文件 。
-T -o 参数将输出 GTF格式。
示例命令:
1.GFF转换GTF
gffread input.gff3 -T -o out.gtf‘
2.GTF转换GFF3
gffread input.gtf -o out.gff3
3.根据GFF或者GTF提取蛋白质,CDS和外显子序列
gffread gene.gff3 -g genome.fa -x cds.fa -y pep.fa -w cdna.fa
三、GFF文件比较
主要采用gffcompare(https://github.com/gpertea/gffcompare),其主要具有三个功能:1)评估Cufflinks/Stringtie等转录本组装软件的准确性;2)合并多个GFF/GTF中重叠的部分(多个样本组装结果的合并)3)可以对一个或多个GTF/GFF文件的注释相对于参考的GTF/GFF文件进行分类(with "class codes" assigned to transcripts as per their relationship with the matching/overlapping reference transcript),如Pacbio预测的GTF与参考GFF比较,修正和评估参考的注释结果。
Usage:
gffcompare [-r
[-o
{-i
常用参数介绍:
-i 多个GTF 文件时,使用此选项较方便,将多个GTF文件写在一个文件中,通过此选项传入即可。
-r 参考的 GTF/GFF文件
-R 针对的是-r参数,仅考虑参考与任何输入的注释文件有重叠的 。
-Q 针对的是-r参数,仅考虑输入的注释文件与任何参考有重叠的 。 (警告,这将丢弃所有的新的注释位点)
-M 丢弃(忽略)掉输入的注释文件和参考注释文件中单外显子转录本
-N 丢弃(忽略)掉参考注释文件中单外显子转录本
-s 基因组序列文件
-e 当评估外显子准确性时,离参考末端外显子最远的距离(默认100)
-d 转录本聚类时起始位点相差的最大距离 (默认100)
-C 在.combined.gtf文件中包含 "contained" 类型的转录本
-F 如果仅是3’端不同,则不丢弃输入的GTF文件中被参考包含的冗余的转录本注释信息。
-G 不丢弃输入的GTF文件中被参考包含的冗余的转录本注释信息,主要是鉴于可变剪接。
-T 对于每一个输入文件不产生 .tmap 和 .refmap文件
-V 给出 GFF 解析时的警告信息
参考命令:
gffcompare -r refChr.gff3 -R -G -o combine input.gtf
输出结果中有以下几个文件:
combine.combined.gtf
combine.loci
combine.stats
combine.tracking
其中在combine.combined.gtf中有一个class_code 代表输入的注释文件与参考注释文件相似性信息,具体如下:
#Transfrag class codes
PriorityCodeDescription
1=Complete match of intron chain
2cContained
3jPotentially novel isoform (fragment): at least one splice junction is shared with a reference transcript
4eSingle exon transfrag overlapping a reference exon and at least 10 bp of a reference intron, indicating a possible pre-mRNA fragment.
5iA transfrag falling entirely within a reference intron
6oGeneric exonic overlap with a reference transcript
7pPossible polymerase run-on fragment (within 2Kbases of a reference transcript)
8rRepeat. Currently determined by looking at the soft-masked reference sequence and applied to transcripts where at least 50% of the bases are lower case
9uUnknown, intergenic transcript
10xExonic overlap with reference on the opposite strand
11sAn intron of the transfrag overlaps a reference intron on the opposite strand (likely due to read mapping errors)
12.(.tracking file only, indicates multiple classifications)
由于输出文件几乎跟cuffcompare格式几乎是一样的,
更详细输出介绍参见http://cole-trapnell-lab.github.io/cufflinks/cuffcompare/。
转自:http://www.huoyunjn.com/wuliuxinwen/2/33709819.htm