随着研究微生物组的需求越来越旺盛,用于微生物组数据分析的平台也越来越多。分享两款最新的微生物组数据分析平台。
01
CoMA
能够处理来自NGS平台的数据,包括Illumina MiSeq,Illumina HiSeq或Illumina NovaSeq,还可以处理以前的454焦磷酸测序技术。CoMA着重于短读而非长读的数据处理。
处理过程包括数据预处理、质量检查、对操作分类单元(OTUs)的聚类、分类、数据后续处理、数据可视化和统计评估。输出结果包含可供发表的图形和标准化格式的文件(例如,制表符分隔的OTU表、biom、Newick tree)。
比起qiime,它的下载安装没那么复杂,兼容linux、mac、windows系统,并且交互式的用户界面更方便小白上手。
性能评估方面,使用了模拟数据和真实的土壤数据与现流行的Mothur、QIIME和QIIME2软件进行了比较,其结果是一致的。
Coma的交互界面
$ coma
如同回答问题般的操作
“Do you want to start a new project?” → Yes
“Do you want to assign your files and choose the number of CPUs?” → Yes
“Are you using paired-end reads?” → Yes
工作流程
使用了各种开源的第三方工具,并以Bash脚本的形式将它们组合到了线性分析工作流程中,从原始输入文件(以FASTQ格式)开始。
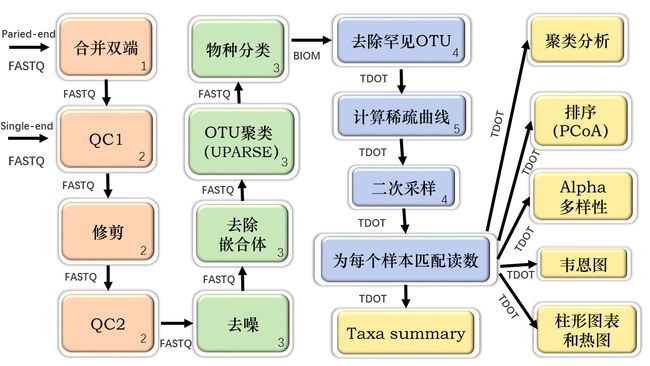
不同的颜色代表CoMA工作流程的四个子部分:数据预处理和质量检查(橙色),OTU的聚类和物种分类(绿色),数据后处理(蓝色)以及数据可视化和统计分析(黄色)。
箭头指示发生的顺序,并指定了每个步骤的输入所需的文件类型。通过BLAST、LAMBDA或RDP使用任一可用数据库完成物种分类(例如Sliva或用户自定义数据库)。
数字表示用于特定CoMA步骤的第三方工具:1 = PANDAseq,2 = PRINSEQ,3 = LotuS / sdm,4 = QIIME,5 = Mothur。TDOT =制表符分隔的OTU表
模拟社区群落数据测试
来自公共模拟微生物库。选择了3个数据集分别为MOCK-13、MOCK-16、MOCK-26.。Mock-13包含21个细菌菌株(18个属;三个重复),MOCK-16包括古细菌,共59个菌株(46属;三个重复),Mock-26包含来自11个真菌菌株(11属)的ITS数据。
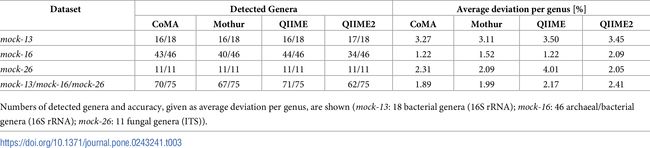
表1展示了四个不同分析平台的基准测试结果:使用CoMA进行的数据分析揭示了18个属中的16个,与Mothur和QIIME数量一致。
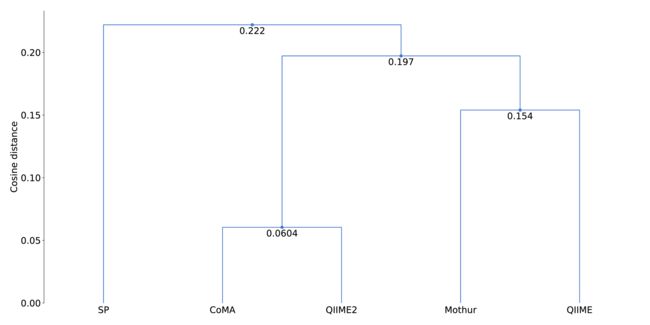
综合所有模拟社区,CoMA表现出总体最高的准确性,每个分类单元的平均偏差为1.89%(表1),而Mothur(1.99%),QIIME(2.17%)和QIIME2(2.41%)。从层次聚类分析来看,CoMA和QIIME2显示最小的余弦距离(0.08)。Mothur的距离为0.11,QIIME的距离为0.12。所有四个管线之间的相互关系比估计值更紧密(总余弦距离:0.18)。
土壤微生物数据集测试
(森林F、草地GR、沼泽S)
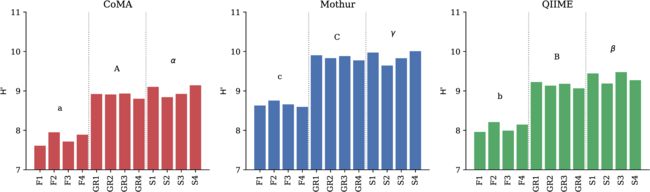
Shannon-Wiener多样性。字母表示不同栖息地使用的分析工具之间的显著差异。三种分析工具都显示出了不同水平的Shannon-Wiener多样性,Mothur>QIIME>CoMA
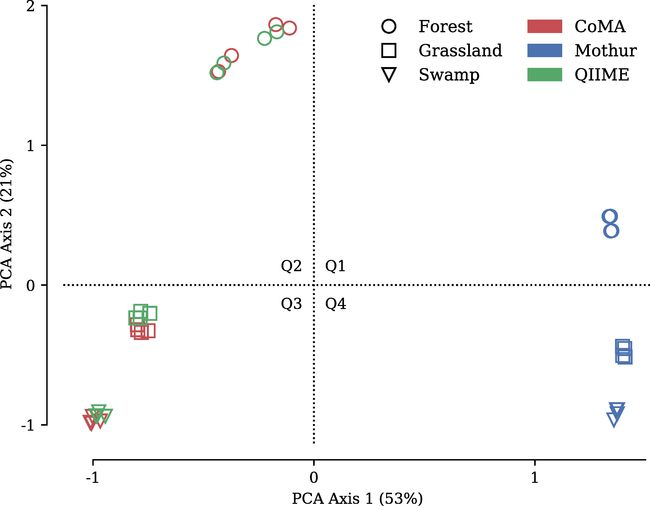
主成分分析结果表示CoMA和QIIME分析得到的微生物群落具有更高的相似性。
韦恩图也显示出相似的结论,无论是哪个分类水平,Mothur分类得到的OTU数目都是最少的,CoMA和QIIME分类得到的一样的OTU的数目最高。
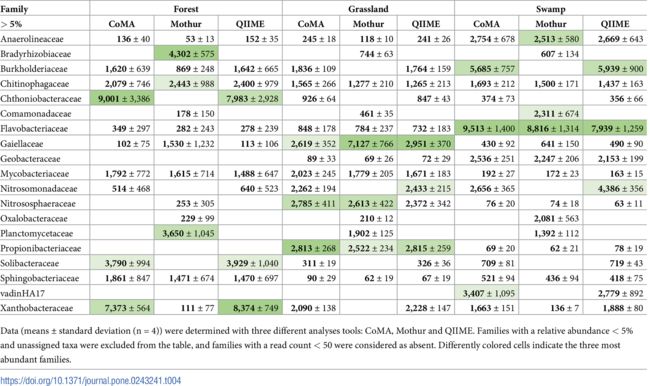
分析了三个土壤类型的关键科水平物种(即丰度>5%的序列)。在表格中被列举出来的关键科水平物种中,无论土壤类型为哪个,三种分析工具都确定了6个关键科。
另一方面,Mothur确定了在CoMA和QIIME中都找不到的4个关键科,CoMA和QIIME再次确定了相同的分类单元(18个中的14个)
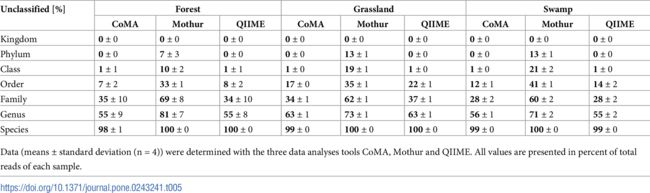
在森林、草原和土壤中的每个分类水平的未分类读数。CoMA和QIIME在所有分类水平上的表现都相似。但是,在分析草地时,CoMA会发现更多的Order和Family(分别为5%和+ 3%; p <0.001)。
总体而言,CoMA的表现与Mothur和QIIME相当,在微生物多样性分析和分类学分类上导致的差异,文章中也做出了相应解释,这通常与算法、参考数据库、参数选择有关。
CoMA的主要优点是图形用户界面支持的直观且用户友好的操作。它使入门级用户无需进行费时费力的培训即可执行扩增子测序数据分析并获得可靠的结果。CoMA还提供了随时停止未完成的分析以稍后再继续或重新计算部分工作流的可能性。通过简单地调整决定性的输入参数,而无需进行完整的重新计算。未来,CoMA将会支持ASV结果输出。
02
MANTA
一款用于研究微生物群与宿主表型数据关系的分析软件,可以本地安装也可在线使用。可以是16SrRNA测序数据,也可以是宏基因组数据。
软件自建了一个微生物组合表型数据的综合数据库,用户能够共享或存储自己的数据,通过数据库和相关分析脚本该可以清楚地展示微生物组和表型数据(例如生活方式等)之间的相关性。依旧是交互式界面,但这个更直观,可以本地安装也可以在线使用。
用户界面
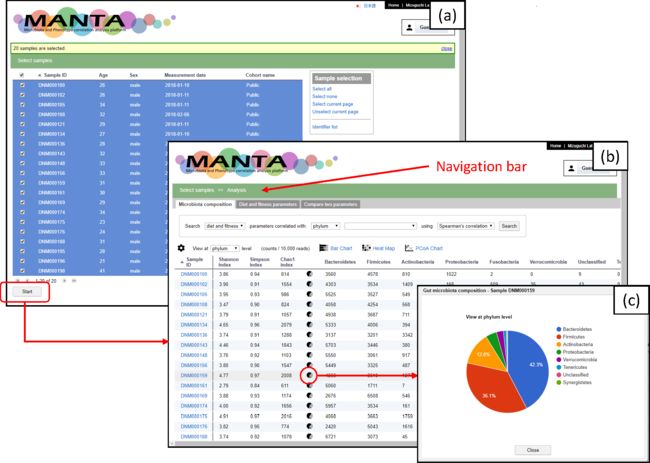
操作类似于MicrobiomeAnalyst这种平台,非常好上手。具体怎么操作就不赘述了,主要看看这款软件在表型数据的分析中表现怎么样。
案例研究
脂肪摄入量与微生物群之间的相关性
数据来源于NIBIOHN队列数据,20名健康成年志愿者(21-41岁,男性)
首先,使用基于OTU的Bray-Curtis距离进行PCoA分析,以评估志愿者之间的相似性。可以通过勾选感兴趣的表型数据进行分组分析,比如图中的abc分别勾选了“食用油”、“脂肪”和“ω-6多不饱和脂肪酸”,然后计算Spearman相关系数,搜索与“脂肪”相关的肠道菌群。
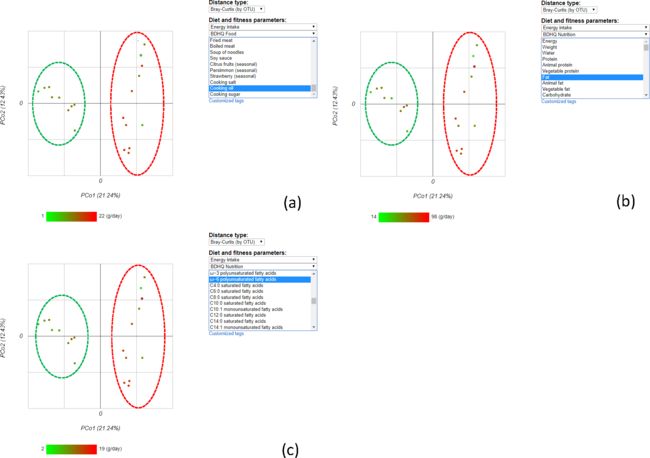
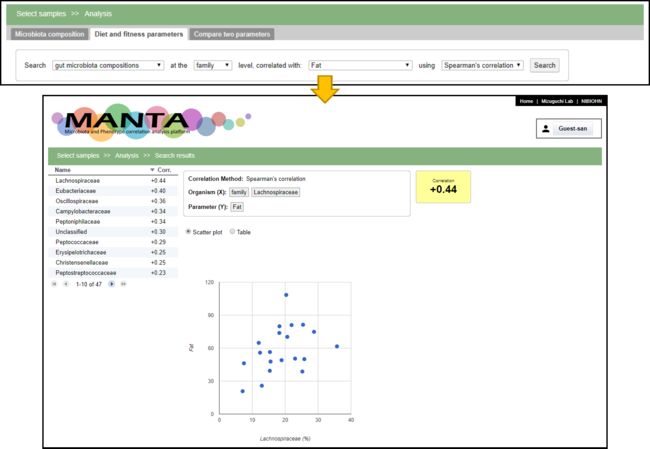
这项分析表明,Lachnospiraceae与脂肪摄入量呈正相关。Lachnospiraceae由丁酸产生菌组成,据研究,低碳水化合物摄入量的高脂肪饮食与Lachnospiraceae和Ruminococcacea的丰度比有关。根据这些结果,推测Lachnospiraceae和Ruminococcacea的比例主要受饮食,特别是脂肪摄入量的影响。
然后反过来,探讨与Lachnospiraceae菌相关的其它表型数据。观察到:
(a)与单不饱和脂肪酸或饱和脂肪酸摄入量呈正相关;
(b)与体育活动时间相关的参数呈负相关;
与Ruminococcaceae菌相关的其它表型数据,观察到:
(c)与ω-6多不饱和脂肪酸摄入量呈正相关;
(d)与体重、总能量消耗、碳水化合物摄入(如煮熟的大米和谷物)等身体成分相关的参数呈负相关。
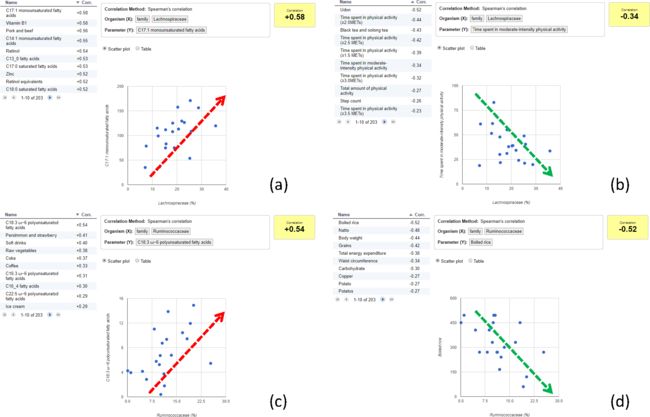
使用MANTA分析出的结果与案例中的结果一致,不同类型的脂肪酸会分别影响Lachnospiraceae和Ruminococcacea。这表示MANTA成功的假设了微生物群和生活方式之间可能存在的联系。
基于以上,我认为MANTA是一个不错的辅助分析工具, MANTA还提供了一个更加简易的版本MANTA BASIC, 输入的文件也比较简单,OUT表和含表型数据的制表符格式文件。
延 伸
软件开发拥有一套复杂的流程。从提出问题,编写方法、开发,然后是一系列的测试,通常包含基准测试、模拟数据的测试、真实的生物数据的测试。每一步都需要经过严格的控制,包括要使用哪些数据,要应用哪种性能指标以及要采用哪种基准。最后整理成文档,将方法放置在软件包或插件中,开放存取,持续的测试更新和版本控制等。复杂但也有逻辑可寻。如果你在开发方面感兴趣,可以阅读这篇文章“Measuring the microbiome: Best practices for developing and benchmarking microbiomics methods”,它详细的介绍了开发用于微生物研究的软件或方法的一套流程,可以说是一篇指南。
03
结 语
工具的选择应该基于解决什么样的问题。对于CoMA,如果你觉得用qiime太困难了,可以试试CoMA,这种非命令式的交互界面还是很好理解的,从案例研究的结果来看,与qiime的结果几乎一致。虽然是新开发的软件,但较为内核的算法借鉴或直接使用了老牌的工具。
MANTA是个研究表型数据与微生物相关性的方便好用的软件,比起自己写代码和调试,这个可以直接上手,很方便。
相关阅读:
微生物多样性测序结果如何看?
宏基因组的一些坑和解决方案