
第一步:创建normal样品的PoN(Create a sites-only PoN with CreateSomaticPanelOfNormals)
To create a panel of normals (PoN), call on each normal sample as if a tumor sample. Then use CreateSomaticPanelOfNormals to output a PoN of germline and artifactual sites. This contrasts with the GATK3 workflow, which uses CombineVariants to retain variant sites called in at least two samples and then uses Picard MakeSitesOnlyVcf to simplify the callset for use as a PoN.
参考:https://software.broadinstitute.org/gatk/documentation/article?id=11136
First, run Mutect2 Mutect2 in tumor-only mode on each normal sample. In tumor-only mode, a single case sample is analyzed with the -tumor flag without an accompanying matched control -normal sample. For the tutorial, we run this command only for sample HG00190.
gatk Mutect2 \
-R ~/Documents/ref/hg38/Homo_sapiens_assembly38.fasta \
-I HG00190.bam \
-tumor HG00190 \
--disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \
-L chr17plus.interval_list \
--germline-resource af-only-gnomad.raw.sites.b37.vcf.gz \
-O 3_HG00190.vcf.gz
Second, collate all the normal VCFs into a single callset with CreateSomaticPanelOfNormals. For the tutorial, to illustrate the step with small data, we run this command on three normal sample VCFs. The general recommendation for panel of normals is a minimum of forty samples.
gatk CreateSomaticPanelOfNormals \
-vcfs 3_HG00190.vcf.gz \
-vcfs 4_NA19771.vcf.gz \
-vcfs 5_HG02759.vcf.gz \
--min-sample-count 2 \
-O 6_threesamplepon.vcf.gz
结果如下:

第二步:call somatic short variants and indel
1、Tumor with matched normal
Given a matched normal, Mutect2 is designed to call somatic variants only. The tool includes logic to skip emitting variants that are clearly present in the germline based on the evidence present in the matched normal. This is done at an early stage to avoid spending computational resources on germline events. If the variant's germline status is borderline, then Mutect2 will emit the variant to the callset with a germline-risk filter. Such filtered emissions enable manual review.
gatk-launch --javaOptions "-Xmx4g" Mutect2 \
-R ref_fasta.fa \
-I tumor.bam \
-tumor tumor_sample_name \
-I normal.bam \
-normal normal_sample_name \
--germline_resource af-only-gnomad.vcf.gz \
--normal_panel pon.vcf.gz \
-L intervals.list \
-O tumor_matched_m2_snvs_indels.vcf.gz
2、Single tumor sample
gatk-launch --javaOptions "-Xmx4g" Mutect2 \
-R ref_fasta.fa \
-I tumor.bam \
-tumor tumor_sample_name \
--germline_resource af-only-gnomad.vcf.gz \
--normal_panel pon.vcf.gz \
-L intervals.list \
-O tumor_unmatched_m2_snvs_indels.vcf.gz
第三步: Estimate cross-sample contamination using GetPileupSummaries and CalculateContamination.
First, run GetPileupSummaries on the tumor BAM to summarize read support for a set number of known variant sites.Use a population germline resource containing only common biallelic variants, e.g. subset by using SelectVariants --restrict-alleles-to BIALLELIC, as well as population AF allele frequencies in the INFO field [4]. The tool tabulates read counts that support reference, alternate and other alleles for the sites in the resource.
gatk GetPileupSummaries \
-I tumor.bam \
-V small_exac_common_3_b37.vcf.gz \
-O 7_tumor_getpileupsummaries.table
This produces a six-column table as shown. The alt_count is the count of reads that support the ALT allele in the germline resource. The allele_frequency corresponds to that given in the germline resource. Counts for other_alt_count refer to reads that support all other alleles.
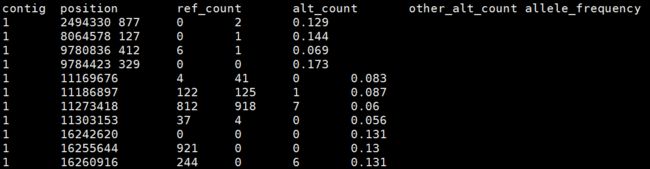
Second, estimate contamination with CalculateContamination. The tool takes the summary table from GetPileupSummaries and gives the fraction contamination. This estimation informs downstream filtering by FilterMutectCalls.
gatk CalculateContamination -I 7_tumor_getpileupsummaries.table -O 8_tumor_calculatecontamination.table
or
gatk CalculateContamination -I 7_tumor_getpileupsummaries.table -matched normal.getpileupsummaries.table -O 8_tumor_calculatecontamination.table
This produces a table with estimates for contamination and error. The estimate for the full tumor sample is shown below and gives a contamination fraction of 0.0205. Going forward, we know to suspect calls with less than ~2% alternate allele fraction.

第四: Filter for confident somatic calls using FilterMutectCalls
FilterMutectCalls determines whether a call is a confident somatic call. The tool uses the annotations within the callset and applies preset thresholds that are tuned for human somatic analyses.
Filter the Mutect2 callset with FilterMutectCalls. Here we use the full callset, somatic_m2.vcf.gz. To activate filtering based on the contamination estimate, provide the contamination table with --contamination-table. In GATK v4.0.0.0, the tool uses the contamination estimate as a hard cutoff.
gatk FilterMutectCalls \
-V somatic_m2.vcf.gz \
--contamination-table tumor_calculatecontamination.table \
-O 9_somatic_oncefiltered.vcf.gz
This produces a VCF callset 9_somatic_oncefiltered.vcf.gz and index. Calls that are likely true positives get the PASS label in the FILTER field, and calls that are likely false positives are labeled with the reason(s) for filtering in the FILTER field of the VCF. We can view the available filters in the VCF header using grep '##FILTER'.
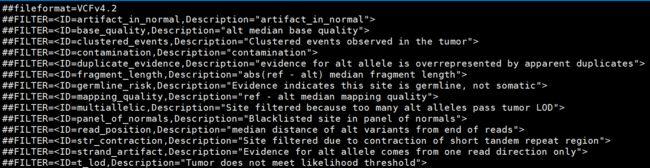

5. (Optional) Estimate artifacts with CollectSequencingArtifactMetrics and filter them with FilterByOrientationBias
FilterByOrientationBias allows filtering based on sequence context artifacts, e.g. OxoG and FFPE. This step is optional and if employed, should always be performed after filtering with FilterMutectCalls. The tool requires the pre_adapter_detail_metricsfrom Picard CollectSequencingArtifactMetrics.
First, collect metrics on sequence context artifacts with CollectSequencingArtifactMetrics. The tool categorizes these as those that occur before hybrid selection (preadapter) and those that occur during hybrid selection (baitbias). Results provide a global view across the genome that empowers decision making in ways that site-specific analyses cannot. The metrics can help decide whether to consider downstream filtering.
gatk CollectSequencingArtifactMetrics \
-I TESTMOS18080001.recal.bam \
-O TESTMOS18080001.artifact \
--FILE_EXTENSION ".txt" \
-R b37.fa
Alternatively, use the tool from a standalone Picard jar.
java -jar picard.jar \
CollectSequencingArtifactMetrics \
I=tumor.bam \
O=10_tumor_artifact \
FILE_EXTENSION=.txt \
R=~/Documents/ref/hg38/Homo_sapiens_assembly38.fasta
This generates five metrics files, including pre_adapter_detail_metrics, which contains counts that FilterByOrientationBias uses. Below are the summary pre_adapter_summary_metrics for the full data. Our samples were not from FFPE so we do not expect this artifact. However, it appears that we could have some OxoG transversions.

Second, perform orientation bias filtering with FilterByOrientationBias. We provide the tool with the once-filtered calls 9_somatic_oncefiltered.vcf.gz, the pre_adapter_detail_metrics file and the sequencing contexts for FFPE (C→T transition) and OxoG (G→T transversion). The tool knows to include the reverse complement contexts.
gatk FilterByOrientationBias \
-A G/T \
-A C/T \
-V 9_somatic_oncefiltered.vcf.gz \
-P tumor_artifact.pre_adapter_detail_metrics.txt \
-O 11_somatic_twicefiltered.vcf.gz
This produces a VCF 11_somatic_twicefiltered.vcf.gz, index and summary 11_somatic_twicefiltered.vcf.gz.summary. In the summary, we see the number of calls for the sequence context and the number of those that the tool filters.
In the VCF header, we see the addition of the 15th filter, orientation_bias, which the tool applies to 56 calls. All 56 of these calls were previously PASS sites, i.e. unfiltered. We now have 673 passing calls out of 3,695 total calls.