NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
单细胞转录组测序发展至今,我们发现许多文章的最后一部分都会落到配受体结合,可是如何挑配受体,哪些基因可能是配受体,我一脸懵逼。。。
于是,我一不小心发现了celltalker(https://arc85.github.io/celltalker/articles/celltalker.html#vignette-overview),大家可以尝试一下哦,嘻嘻,废话不多说。
Introduction
对单细胞RNAseq数据可能进行的多种分析之一是评估细胞间的交流 (cell-cell communication)。celltalker通过寻找细胞群内和细胞群之间已知的配体和受体对的表达来评估细胞之间的交流。 我们采用的配受体数据库来自Ramilowski等人于2015年在Nature communication上发表的A draft network of ligand-receptor-mediated multicellular signalling in human描述的一组配体和受体。我们建议使用此数据集作为起点,并整理自己的已知配体和受体列表。另外Tormo 2018年发表的Nature文章Single-cell reconstruction of the early maternal-fetal interface in humans扩展了受体和配体对,也会应用于cellTalker的更新版中。
为了获得可靠的结果,我们要求每个组中都有多个重复样品,并且只对不同组间一致性表达的配体和受体感兴趣(而仅在单个重复中发现的互作可信度低)。我们通过评估每组中各个重复的表达矩阵并仅对满足一定阈值(这个阈值随意性也比较强)的相互作用进行提取。
配体和受体相互作用的差异至少在三种方面具有生物学意义:
- 在一组细胞中独特地存在;
- 各个cluster间配体或受体的互作差异;
- 参与组间配体和受体相互作用的细胞网络的差异。
我们提供了评估每种潜在生物学差异的方法,并提供了具体示例。
在这个vignette中,我们展示了cellTalker在评估健康捐献者外周血(N = 2)和扁桃体(N = 3)中鉴定配体/受体相互作用的基本应用。该数据可从我们最近发布的数据集GSE139324中获得 (Cillo et al, Immunity 2020)。
Vignette overview
展示Celltalker应用于10X Genomics数据的的标准用法。具体分为下面几步:
- 使用标准的Seurat工作流程(v.3.1.1)对数据进行聚类;
- 使用Celltalker建立样品组中稳定表达的配体和受体的列表;
- 确定配体/受体相互作用;
- 评估组之间特异表达的配体/受体对;
- 识别和可视化组特异的配体/受体对;
Installation
library(devtools)
install_github("arc85/celltalker")
library(celltalker)
Clustering data with Seurat
使用Seurat进行标准的聚类分析和免疫谱系识别(假设已从GEO下载了raw matrix)。(重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))
suppressMessages({
library(Seurat)
library(celltalker)
})
# 设置可重复性的种子数字
set.seed(02221989)
# 读取raw data
# path.to.working:自行修改路径
path.to.working <- “”
setwd(paste(path.to.working,"/data_matrices/",sep=""))
data.paths <- list.files()
# GRCh38 根据需要调整为其它基因组版本
specific.paths <- paste(path.to.working,"data_matrices",data.paths,"GRCh38",sep="/")
setwd(path.to.working)
raw.data <- Read10X(specific.paths)
# 设置metadata (这一部分是这个数据特异的,实际还是自己整理个metadata文件更为方便)
sample.data <- data.frame(matrix(data=NA,nrow=ncol(raw.data),ncol=2))
rownames(sample.data) <- colnames(raw.data)
colnames(sample.data) <- c("sample.id","sample.type")
sample.data[grep("^[A-z]",rownames(sample.data)),"sample.id"] <- "pbmc_1"
sample.data[grep("^2",rownames(sample.data)),"sample.id"] <- "tonsil_1"
sample.data[grep("^3",rownames(sample.data)),"sample.id"] <- "pbmc_2"
sample.data[grep("^4",rownames(sample.data)),"sample.id"] <- "tonsil_2"
sample.data[grep("^5",rownames(sample.data)),"sample.id"] <- "pbmc_3"
sample.data[grep("^6",rownames(sample.data)),"sample.id"] <- "tonsil_3"
sample.data[,"sample.type"] <- sapply(strsplit(sample.data$sample.id,split="_"),function(x) x[1])
## 使用原始数据创建Seurat对象,并添加sample-specific metadata
ser.obj <- CreateSeuratObject(counts=raw.data,meta.data=sample.data)
Seurat三部曲
#标准Seurat工作流程
ser.obj <- NormalizeData(ser.obj)
ser.obj <- FindVariableFeatures(ser.obj)
ser.obj <- ScaleData(ser.obj)
PCA分析
ser.obj <- RunPCA(ser.obj)
获得对各个主成分贡献比较大的基因 (用了这么多年的PCA可视化竟然是错的!!!)
## PC_ 1
## Positive: S100A6, IL32, S100A4, ANXA1, VIM, FTL, TRBC1, SRGN, S100A9, S100A8
## TYROBP, LYZ, CTSW, XIST, NEAT1, VCAN, S100A12, FCER1G, S100A11, FCN1
## PLAC8, ID2, CCL5, NKG7, CST3, CSTA, ZFP36, IL1B, MT2A, KLRB1
## Negative: RGS13, KIAA0101, NUSAP1, AURKB, MKI67, BIRC5, TYMS, TOP2A, TK1, CDKN3
## UBE2C, PTTG1, CDK1, STMN1, CCNB2, GTSE1, BIK, RRM2, TCL1A, SHCBP1
## CDCA3, CDC20, TPX2, LRMP, CCNA2, MND1, CCNB1, PBK, ZWINT, RMI2
## PC_ 2
## Positive: CST3, LYZ, FCN1, CSTA, S100A9, S100A8, TYROBP, LST1, FGL2, VCAN
## S100A12, SERPINA1, MNDA, FCER1G, CLEC7A, MS4A6A, CD14, CFD, IL1B, TYMP
## LGALS1, RP11-1143G9.4, AIF1, CTSS, NAMPT, CFP, TNFSF13B, CSF3R, MPEG1, TMEM176B
## Negative: IL32, NPM1, CD69, TRBC1, ISG20, ITM2A, IGKC, IGHA1, HSP90AB1, DDIT4
## HIST1H4C, PSIP1, AQP3, MYC, PIM2, HMGN1, PASK, NUCB2, HSPA1B, HSPB1
## CD79A, SUSD3, KLRB1, SYNE2, CHI3L2, IGHG3, IGLC2, FKBP11, IGHG1, SH2D1A
## PC_ 3
## Positive: IL32, NKG7, CTSW, TRBC1, GZMA, CST7, GNLY, MKI67, ANXA1, TOP2A
## CCL5, PRF1, BIRC5, S100A4, KLRB1, CCNA2, AURKB, CENPF, GTSE1, CDKN3
## KLRD1, UBE2C, CDK1, TYMS, TPX2, RRM2, ID2, S100A6, FGFBP2, CDC20
## Negative: HLA-DRA, HLA-DQA1, HLA-DQB1, CD79A, HLA-DRB1, MS4A1, CD74, HLA-DPA1, HLA-DPB1, CD79B
## HLA-DMA, HLA-DMB, BANK1, VPREB3, IGKC, HLA-DRB5, MEF2C, CD22, IRF8, CD19
## SMIM14, FCRLA, HLA-DOB, CD24, CD40, FCER2, BLK, HLA-DQA2, IGHD, CTSH
## PC_ 4
## Positive: TOP2A, UBE2C, MKI67, GTSE1, CENPF, AURKB, PLK1, CCNA2, CDK1, CDCA8
## HMMR, CDCA3, CDC20, TPX2, CDKN3, DLGAP5, CENPE, BIRC5, CCNB2, CENPA
## KIF2C, CKAP2L, PBK, NUSAP1, KIFC1, AURKA, SPC25, NUF2, KIF23, ASPM
## Negative: NKG7, GNLY, CST7, GZMB, GZMA, PRF1, KLRD1, FGFBP2, CCL5, KLRF1
## HOPX, CTSW, GZMH, TRDC, FCGR3A, SPON2, CLIC3, MATK, ADGRG1, S1PR5
## CCL4, CMC1, XCL2, PFN1, CD160, FCRL6, IL2RB, TRGC1, KLRC1, C12orf75
## PC_ 5
## Positive: ICA1, PDCD1, TBC1D4, ITM2A, ICOS, MAF, TOX2, IL32, TNFRSF4, PASK
## PKM, SMCO4, ACTG1, CORO1B, CTLA4, NPM1, TRBC1, PCAT29, TIGIT, AC133644.2
## TOX, ANP32B, ENO1, GBP2, COTL1, GAPDH, SUSD3, PIM2, AQP3, SERPINA9
## Negative: NKG7, GNLY, KLRD1, FGFBP2, GZMB, GZMA, KLRF1, CCL5, PRF1, TRDC
## GZMH, CST7, CTSW, BANK1, MATK, PLK1, HMMR, HLA-DPB1, CENPA, CLIC3
## GTSE1, CENPE, CCL4, SPON2, PDLIM1, HLA-DPA1, CDCA8, DLGAP5, TPX2, IGHD
拐点法寻找top可用的主成分用于后续分析 (具体选择方式见:(重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)))
ElbowPlot(ser.obj)
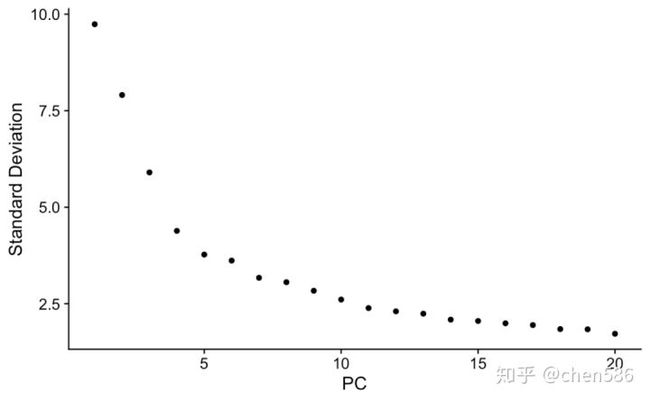
降维可视化
#我们选择top 15 PCs用于后续分析
ser.obj <- RunUMAP(ser.obj,reduction="pca", dims=1:15)
聚类
## Computing nearest neighbor graph
ser.obj <- FindNeighbors(ser.obj,reduction="pca",dims=1:15)
## Computing SNN
ser.obj <- FindClusters(ser.obj,resolution=0.5)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 15524
## Number of edges: 543084
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.9185
## Number of communities: 17
## Elapsed time: 2 seconds
画图看一看!ggplot2高效实用指南 (可视化脚本、工具、套路、配色)
p1 <- DimPlot(ser.obj,reduction="umap",group.by="sample.id")
p2 <- DimPlot(ser.obj,reduction="umap",group.by="sample.type")
p3 <- DimPlot(ser.obj,reduction="umap",group.by="RNA_snn_res.0.5",label=T) + NoLegend()
cowplot::plot_grid(p1,p2,p3)
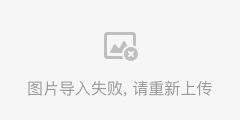
让我们看看部分基因的表达情况!
FeaturePlot(ser.obj,reduction="umap",features=c("CD3D","CD8A","CD4","CD14","MS4A1","FCGR3A","IL3RA"))
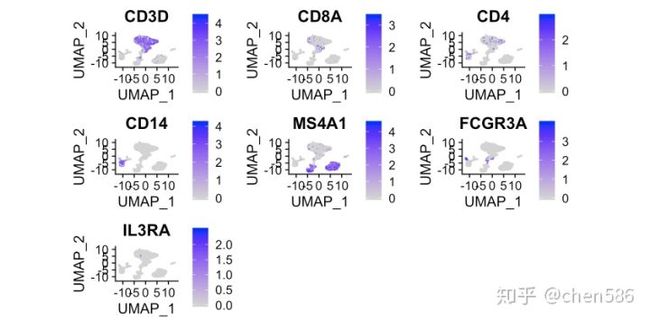
命名细胞簇并移除红细胞 (cellassign:用于肿瘤微环境分析的单细胞注释工具(9月Nature))
# 为细胞类型增加metadata
cell.types <- vector("logical",length=ncol(ser.obj))
names(cell.types) <- colnames(ser.obj)
cell.types[[email protected]$RNA_snn_res.0.5=="0"] <- "cd4.tconv"
cell.types[[email protected]$RNA_snn_res.0.5=="1"] <- "cd4.tconv"
cell.types[[email protected]$RNA_snn_res.0.5=="2"] <- "b.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="3"] <- "b.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="4"] <- "cd14.monocytes"
cell.types[[email protected]$RNA_snn_res.0.5=="5"] <- "cd8.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="6"] <- "cd4.tconv"
cell.types[[email protected]$RNA_snn_res.0.5=="7"] <- "cd4.tconv"
cell.types[[email protected]$RNA_snn_res.0.5=="8"] <- "b.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="9"] <- "b.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="10"] <- "nk.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="11"] <- "cd8.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="12"] <- "plasma.cells"
cell.types[[email protected]$RNA_snn_res.0.5=="13"] <- "cd14.monocytes"
cell.types[[email protected]$RNA_snn_res.0.5=="14"] <- "cd16.monocytes"
cell.types[[email protected]$RNA_snn_res.0.5=="15"] <- "pdc"
cell.types[[email protected]$RNA_snn_res.0.5=="16"] <- "RBCs"
ser.obj[["cell.types"]] <- cell.types
#去除红细胞
rbc.cell.names <- names(cell.types)[[email protected]$RNA_snn_res.0.5=="16"]
ser.obj <- ser.obj[,!colnames(ser.obj) %in% rbc.cell.names]
Consistently expressed ligands and receptors
现在,我们已经在数据中识别并命名了cluster,我们将继续进行celltalker分析。随该软件包一起提供的有一个ramilowski_pairs,它是一个由配体、受体和推测的配体受体对组成的data.frame。
首先,根据通用型配体和受体从我们的数据集中选出对应的基因,然后进行差异基因分析,只保留在样品组之间差异表达的配体受体。
然后,我们将为每个重复样本创建单独的数据矩阵,存储为tibble格式,以便于使用tidyverse进行后续处理。
(生信宝典注:如果我们自己有受体配体对,也可以整理成这样一个三列的格式,导入进来,替换掉数据包原有的配体受体对信息,实现更加定制的分析。)
#查阅 ramilowski_pairs 数据集
head(ramilowski_pairs)
## ligand receptor pair
## 1 A2M LRP1 A2M_LRP1
## 2 AANAT MTNR1A AANAT_MTNR1A
## 3 AANAT MTNR1B AANAT_MTNR1B
## 4 ACE AGTR2 ACE_AGTR2
## 5 ACE BDKRB2 ACE_BDKRB2
## 6 ADAM10 AXL ADAM10_AXL
dim(ramilowski_pairs)
## [1] 2557 3
#该数据集中有2,557个特异的配体/受体对
#鉴定差异表达的配体和受体
#在我们的数据集中识别配体和受体
ligs <- as.character(unique(ramilowski_pairs$ligand))
recs <- as.character(unique(ramilowski_pairs$receptor))
ligs.present <- rownames(ser.obj)[rownames(ser.obj) %in% ligs]
recs.present <- rownames(ser.obj)[rownames(ser.obj) %in% recs]
genes.to.use <- union(ligs.present,recs.present) # union用于合并子集
# 使用FindAllMarkers区分组之间差异表达的配体和受体
Idents(ser.obj) <- "sample.type"
markers <- FindAllMarkers(ser.obj, assay="RNA",features=genes.to.use,only.pos=TRUE)
ligs.recs.use <- unique(markers$gene)
length(ligs.recs.use)
## [1] 61
#产生61个配体和受体以进行评估
#过滤ramilowski配受对
interactions.forward1 <- ramilowski_pairs[as.character(ramilowski_pairs$ligand) %in% ligs.recs.use,]
interactions.forward2 <- ramilowski_pairs[as.character(ramilowski_pairs$receptor) %in% ligs.recs.use,]
interact.for <- rbind(interactions.forward1, interactions.forward2)
dim(interact.for)
## [1] 241 3
生成celltalker的输入数据
#产生241个配体和受体相互作用
#Create data for celltalker
expr.mat <- GetAssayData(ser.obj,slot="counts")
defined.clusters <- [email protected]$cell.types
defined.groups <- [email protected]$sample.type
defined.replicates <- [email protected]$sample.id
reshaped.matrices <- reshape_matrices(count.matrix=expr.mat,clusters=defined.clusters,groups=defined.groups,replicates=defined.replicates,ligands.and.receptors=interact.for)
#查看tibble的层次结构
reshaped.matrices
## # A tibble: 2 x 2
## group samples
##
## 1 pbmc
## 2 tonsil
数据展开为单个样品展示
unnest(reshaped.matrices,cols="samples")
## # A tibble: 6 x 3
## group sample expr.matrices
##
## 1 pbmc pbmc_1
## 2 pbmc pbmc_2
## 3 pbmc pbmc_3
## 4 tonsil tonsil_1
## 5 tonsil tonsil_2
## 6 tonsil tonsil_3
names(pull(unnest(reshaped.matrices,cols="samples"))[[1]])
## [1] "b.cells" "cd14.monocytes" "cd16.monocytes" "cd4.tconv"
## [5] "cd8.cells" "nk.cells" "pdc" "plasma.cells"
在此初始步骤中,我们要做的是将我们的整体表达矩阵中给每个样本中分离出单独的表达矩阵。
接下来,使用create_lig_rec_tib函数为每个组创建一组一致表达的配体和受体。
# cells.reqd=10:每个cluster中至少有10个细胞表达了配体/受体
# freq.pos.reqd=0.5:至少有50%重复个体中表达的配体/受体
consistent.lig.recs <- create_lig_rec_tib(exp.tib=reshaped.matrices,clusters=defined.clusters,groups=defined.groups,replicates=defined.replicates,cells.reqd=10,freq.pos.reqd=0.5,ligands.and.receptors=interact.for)
consistent.lig.recs
## # A tibble: 2 x 2
## group lig.rec.exp
##
## 1 pbmc
## 2 tonsil
unnest(consistent.lig.recs[1,2],cols="lig.rec.exp")
## # A tibble: 8 x 2
## cluster.id ligands.and.receptors
##
## 1 b.cells
## 2 cd14.monocytes
## 3 cd16.monocytes
## 4 cd4.tconv
## 5 cd8.cells
## 6 nk.cells
## 7 pdc
## 8 plasma.cells
pull(unnest(consistent.lig.recs[1,2],cols="lig.rec.exp")[1,2])[[1]]
根据上面指定的标准,我们现在已经从每个实验组的每个cluster中获得了一致表达的配体和受体的列表。
## $ligands
## [1] "S100A9" "S100A8" "IL1B" "FN1" "BTLA" "SPON2"
## [7] "LRPAP1" "VCAN" "CD14" "LY86" "HLA-G" "HLA-A"
## [13] "HLA-E" "HLA-B" "LTB" "HSPA1A" "CD24" "NAMPT"
## [19] "TIMP1" "CD40LG" "ADAM28" "PNOC" "IL7" "ANXA1"
## [25] "SEMA4D" "VIM" "PSAP" "LYZ" "SELPLG" "HMGB1"
## [31] "TNFSF13B" "GZMB" "CALM1" "SERPINA1" "HSP90AA1" "B2M"
## [37] "PKM" "IL16" "CCL5" "CCL3" "ICAM2" "CD70"
## [43] "ICAM1" "ICAM3" "TGFB1" "FLT3LG" "APP"
##
## $receptors
## [1] "CSF3R" "TGFBR3" "KCNA3" "CD53" "CD1A" "CD247"
## [7] "SELL" "CXCR4" "ITGA4" "GRM7" "TGFBR2" "CCR5"
## [13] "TFRC" "TLR1" "IL7R" "CD180" "ADRB2" "CD74"
## [19] "HMMR" "HLA-F" "KCNQ5" "IGF2R" "CCR6" "CD36"
## [25] "CXCR3" "PGRMC1" "CD72" "TGFBR1" "ABCA1" "IFITM1"
## [31] "CD81" "KCNQ1" "CD44" "CD82" "IL10RA" "CD3D"
## [37] "CD3G" "CXCR5" "SORL1" "APLP2" "ITGB1" "FAS"
## [43] "CD27" "CD4" "KLRG1" "CLEC2B" "KLRD1" "KLRC1"
## [49] "PDE1B" "CD63" "TNFRSF17" "CD19" "ITGAL" "ITGAM"
## [55] "TNFRSF13B" "ERBB2" "PTPRA" "CD40" "OPRL1" "INSR"
## [61] "TYROBP" "CD79A" "KCNN4" "FPR2" "LILRB2" "LILRB1"
## [67] "KIR2DL3" "KIR2DL1" "KIR3DL2" "TNFRSF13C" "CELSR1" "ITGB2"
Determine putative ligand/receptor pairs
获得稳定表达的受体-配体后,鉴定给定样品组细胞簇内和细胞簇间可能存在的互作 (基于ramilowski_pairs$pair数据)。获得的Tibble数据 (put.int)中包含样本组和每个组的配体/受体对列表,以及参与配体/受体相互作用的cluster。
# freq.group.in.cluster: 只对包含细胞数大于总细胞数5%的簇进行互作分析
put.int <- putative_interactions(ligand.receptor.tibble=consistent.lig.recs,clusters=defined.clusters,groups=defined.groups,freq.group.in.cluster=0.05,ligands.and.receptors=interact.for)
## Warning: `cols` is now required.
## Please use `cols = c(lig.rec.exp)`
## Warning: `cols` is now required.
## Please use `cols = c(lig.rec.exp)`
Identifying and visualizing unique ligand/receptor pairs in a group
现在我们有了配体/受体相互作用的列表,我们可以使用unique_interactions函数鉴定组特异的互作,并使用circos_plot函数可视化组之间的差异。
#Identify unique ligand/receptor interactions present in each sample
unique.ints <- unique_interactions(put.int,group1="pbmc",group2="tonsil",interact.for)
#Get data to plot circos for PBMC
pbmc.to.plot <- pull(unique.ints[1,2])[[1]]
for.circos.pbmc <- pull(put.int[1,2])[[1]][pbmc.to.plot]
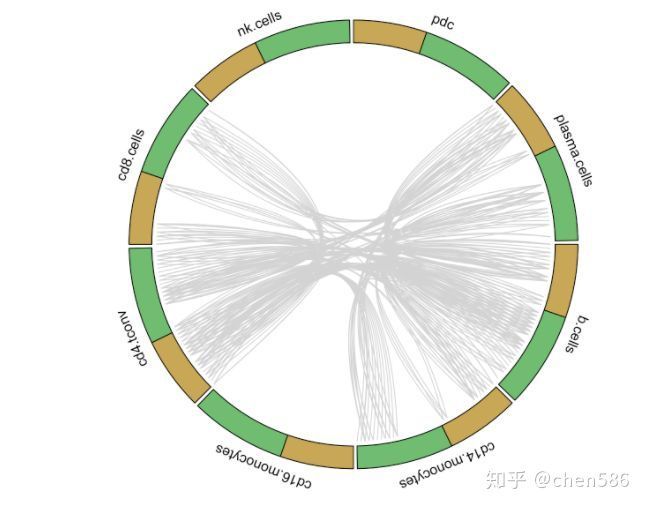
circos_plot(interactions=for.circos.pbmc,clusters=defined.clusters)
PBMC组特有的受体-配体互作
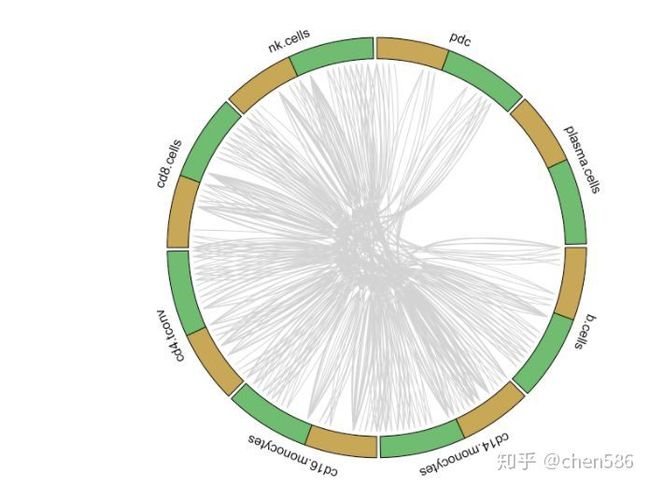
从以上图中我们可以看出研究人员用黄色表示配体基因,绿色表示受体基因,然后将可以相互配对的基因连在一起构成簇之间的互作关系。
Tonsil组特有的受体-配体互作
#Get data to plot circos for tonsil
tonsil.to.plot <- pull(unique.ints[2,2])[[1]]
for.circos.tonsil <- pull(put.int[2,2])[[1]][tonsil.to.plot]
circos_plot(interactions=for.circos.tonsil,clusters=defined.clusters)
Tonsil组特有的受体-配体互作
- CIRCOS圈图绘制 - circos安装
- CIRCOS圈图绘制 - 最简单绘图和解释
- CIRCOS圈图绘制 - 染色体信息展示和调整
- CIRCOS增加热图、点图、线图和区块属性
作者:May(协和医院)
编辑:生信宝典