
https://doi.org/10.15252/msb.20209923
Abstract
Molecular knowledge of biological processes is a cornerstone in omics data analysis. Applied to single‐cell data, such analyses provide mechanistic insights into individual cells and their interactions. However, knowledge of intercellular communication is scarce, scattered across resources, and not linked to intracellular processes. To address this gap, we combined over 100 resources covering interactions and roles of proteins in inter‐ and intracellular signaling, as well as transcriptional and post‐transcriptional regulation. We added protein complex information and annotations on function, localization, and role in diseases for each protein. The resource is available for human, and via homology translation for mouse and rat. The data are accessible via OmniPath’s web service (https://omnipathdb.org/), a Cytoscape plug‐in, and packages in R/Bioconductor and Python, providing access options for computational and experimental scientists. We created workflows with tutorials to facilitate the analysis of cell–cell interactions and affected downstream intracellular signaling processes. OmniPath provides a single access point to knowledge spanning intra‐ and intercellular processes for data analysis, as we demonstrate in applications studying SARS‐CoV‐2 infection and ulcerative colitis.
what:背景:关于细胞间通讯的知识很少,并且与细胞内过程没有联系。
Pypath包含所有使用的资源的URL,包括标识符转换表。 它会为每个操作自动下载,提取和预处理数据。 然后,它将下载的数据存储在属于计算机上用户帐户的本地缓存目录中。 创建缓存后,pypath会从中读取并仅在用户请求时执行下载。
利用正常和感染的人类肺细胞系的RNA-seq数据分析差异基因,选择过表达的配体?
基因集富集分析,选择炎症反应基因作为target
nichenetr :选择受体和转录因子之间的最短路径
covid-19胞内和胞外的信号相互作用
ulcerative colitis 溃疡性结肠炎

https://www.nature.com/articles/s41421-020-0153-3
Abstract
Human coronaviruses (HCoVs), including severe acute respiratory syndrome coronavirus (SARS-CoV) and 2019 novel coronavirus (2019-nCoV, also known as SARS-CoV-2), lead global epidemics with high morbidity and mortality. However, there are currently no effective drugs targeting 2019-nCoV/SARS-CoV-2. Drug repurposing, representing as an effective drug discovery strategy from existing drugs, could shorten the time and reduce the cost compared to de novo drug discovery. In this study, we present an integrative, antiviral drug repurposing methodology implementing a systems pharmacology-based network medicine platform, quantifying the interplay between the HCoV–host interactome and drug targets in the human protein–protein interaction network. Phylogenetic analyses of 15 HCoV whole genomes reveal that 2019-nCoV/SARS-CoV-2 shares the highest nucleotide sequence identity with SARS-CoV (79.7%). Specifically, the envelope and nucleocapsid proteins of 2019-nCoV/SARS-CoV-2 are two evolutionarily conserved regions, having the sequence identities of 96% and 89.6%, respectively, compared to SARS-CoV. Using network proximity analyses of drug targets and HCoV–host interactions in the human interactome, we prioritize 16 potential anti-HCoV repurposable drugs (e.g., melatonin, mercaptopurine, and sirolimus) that are further validated by enrichment analyses of drug-gene signatures and HCoV-induced transcriptomics data in human cell lines. We further identify three potential drug combinations (e.g., sirolimus plus dactinomycin, mercaptopurine plus melatonin, and toremifene plus emodin) captured by the “Complementary Exposure” pattern: the targets of the drugs both hit the HCoV–host subnetwork, but target separate neighborhoods in the human interactome network. In summary, this study offers powerful network-based methodologies for rapid identification of candidate repurposable drugs and potential drug combinations targeting 2019-nCoV/SARS-CoV-2.
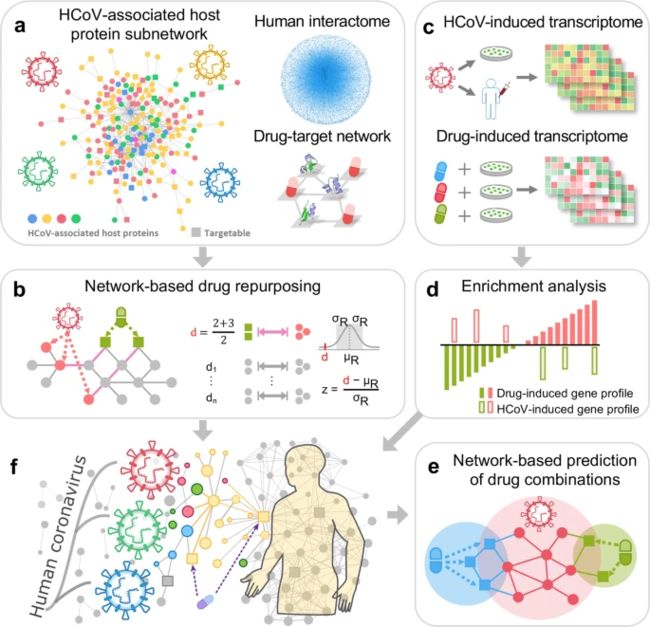
covid-19的药物重定位
量化人类蛋白质-蛋白质相互作用网络中HCoV-宿主相互作用组和药物靶点之间相互作用。
人体相互作用组中药物靶标和HCoV-宿主相互作用的网络邻近性分析
validated by enrichment analyses of drug-gene signatures and HCoV-induced transcriptomics data in human cell lines.
the targets of the drugs both hit the HCoV–host subnetwork, but target separate neighborhoods in the human interactome network.

https://www.thelancet.com/journals/landig/article/PIIS2589-7500(20)30192-8/fulltext#seccestitle70
Summary
Drug repurposing or repositioning is a technique whereby existing drugs are used to treat emerging and challenging diseases, including COVID-19. Drug repurposing has become a promising approach because of the opportunity for reduced development timelines and overall costs. In the big data era, artificial intelligence (AI) and network medicine offer cutting-edge application of information science to defining disease, medicine, therapeutics, and identifying targets with the least error. In this Review, we introduce guidelines on how to use AI for accelerating drug repurposing or repositioning, for which AI approaches are not just formidable but are also necessary. We discuss how to use AI models in precision medicine, and as an example, how AI models can accelerate COVID-19 drug repurposing. Rapidly developing, powerful, and innovative AI and network medicine technologies can expedite therapeutic development. This Review provides a strong rationale for using AI-based assistive tools for drug repurposing medications for human disease, including during the COVID-19 pandemic.

how使用AI加速药物发现:
FNN
CNN
RNN

https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.6b00248
Abstract
Deep learning is rapidly advancing many areas of science and technology with multiple success stories in image, text, voice and video recognition, robotics, and autonomous driving. In this paper we demonstrate how deep neural networks (DNN) trained on large transcriptional response data sets can classify various drugs to therapeutic categories solely based on their transcriptional profiles. We used the perturbation samples of 678 drugs across A549, MCF-7, and PC-3 cell lines from the LINCS Project and linked those to 12 therapeutic use categories derived from MeSH. To train the DNN, we utilized both gene level transcriptomic data and transcriptomic data processed using a pathway activation scoring algorithm, for a pooled data set of samples perturbed with different concentrations of the drug for 6 and 24 hours. In both pathway and gene level classification, DNN achieved high classification accuracy and convincingly outperformed the support vector machine (SVM) model on every multiclass classification problem, however, models based on pathway level data performed significantly better. For the first time we demonstrate a deep learning neural net trained on transcriptomic data to recognize pharmacological properties of multiple drugs across different biological systems and conditions. We also propose using deep neural net confusion matrices for drug repositioning. This work is a proof of principle for applying deep learning to drug discovery and development.

数据:利用转录组数据
pathwary activation score:信号通路的激活,降维
perturbation samples: perturbation time, perturbation concentration, and cell line parameters

https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbab115/6224261#235073573
Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), better known as COVID-19, has become a current threat to humanity. The second wave of the SARS-CoV-2 virus has hit many countries, and the confirmed COVID-19 cases are quickly spreading. Therefore, the epidemic is still passing the terrible stage. Having idiopathic pulmonary fibrosis (IPF) and chronic obstructive pulmonary disease (COPD) are the risk factors of the COVID-19, but the molecular mechanisms that underlie IPF, COPD, and CVOID-19 are not well understood. Therefore, we implemented transcriptomic analysis to detect common pathways and molecular biomarkers in IPF, COPD, and COVID-19 that help understand the linkage of SARS-CoV-2 to the IPF and COPD patients. Here, three RNA-seq datasets (GSE147507, GSE52463, and GSE57148) from Gene Expression Omnibus (GEO) is employed to detect mutual differentially expressed genes (DEGs) for IPF, and COPD patients with the COVID-19 infection for finding shared pathways and candidate drugs. A total of 65 common DEGs among these three datasets were identified. Various combinatorial statistical methods and bioinformatics tools were used to build the protein–protein interaction (PPI) and then identified Hub genes and essential modules from this PPI network. Moreover, we performed functional analysis under ontologies terms and pathway analysis and found that IPF and COPD have some shared links to the progression of COVID-19 infection. Transcription factors–genes interaction, protein–drug interactions, and DEGs-miRNAs coregulatory network with common DEGs also identified on the datasets. We think that the candidate drugs obtained by this study might be helpful for effective therapeutic in COVID-19.

识别SARS-CoV-2感染对特发性肺纤维化和慢性阻塞性肺疾病患者的影响的生物信息学和系统生物学方法
背景:特发性肺纤维化和慢性阻塞性肺疾病是covid-19的风险因子,但是他们三者之间的分子机制尚不清楚
what?这三者之间的共享通路和分子标志物
how?
protein-drug:假设:共享差异基因是潜在的药物靶点 Enrichr

https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000970
Abstract
The global coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to unprecedented social and economic consequences. The risk of morbidity and mortality due to COVID-19 increases dramatically in the presence of coexisting medical conditions, while the underlying mechanisms remain unclear. Furthermore, there are no approved therapies for COVID-19. This study aims to identify SARS-CoV-2 pathogenesis, disease manifestations, and COVID-19 therapies using network medicine methodologies along with clinical and multi-omics observations. We incorporate SARS-CoV-2 virus–host protein–protein interactions, transcriptomics, and proteomics into the human interactome. Network proximity measurement revealed underlying pathogenesis for broad COVID-19-associated disease manifestations. Analyses of single-cell RNA sequencing data show that co-expression of ACE2 and TMPRSS2 is elevated in absorptive enterocytes from the inflamed ileal tissues of Crohn disease patients compared to uninflamed tissues, revealing shared pathobiology between COVID-19 and inflammatory bowel disease. Integrative analyses of metabolomics and transcriptomics (bulk and single-cell) data from asthma patients indicate that COVID-19 shares an intermediate inflammatory molecular profile with asthma (including IRAK3 and ADRB2). To prioritize potential treatments, we combined network-based prediction and a propensity score (PS) matching observational study of 26,779 individuals from a COVID-19 registry. We identified that melatonin usage (odds ratio [OR] = 0.72, 95% CI 0.56–0.91) is significantly associated with a 28% reduced likelihood of a positive laboratory test result for SARS-CoV-2 confirmed by reverse transcription–polymerase chain reaction assay. Using a PS matching user active comparator design, we determined that melatonin usage was associated with a reduced likelihood of SARS-CoV-2 positive test result compared to use of angiotensin II receptor blockers (OR = 0.70, 95% CI 0.54–0.92) or angiotensin-converting enzyme inhibitors (OR = 0.69, 95% CI 0.52–0.90). Importantly, melatonin usage (OR = 0.48, 95% CI 0.31–0.75) is associated with a 52% reduced likelihood of a positive laboratory test result for SARS-CoV-2 in African Americans after adjusting for age, sex, race, smoking history, and various disease comorbidities using PS matching. In summary, this study presents an integrative network medicine platform for predicting disease manifestations associated with COVID-19 and identifying melatonin for potential prevention and treatment of COVID-19.
一种网络医学方法,用于调查和基于人群的疾病表现验证以及针对COVID-19的药物替代用途。
背景:这项研究旨在使用网络医学方法以及临床和多组学观察来确定SARS-CoV-2的发病机制,疾病表现和COVID-19治疗。
what: We incorporate SARS-CoV-2 virus–host protein–protein interactions, transcriptomics, and proteomics into the human interactome.
网络邻近度测量揭示了广泛的COVID-19相关疾病表现的潜在发病机理。
1.疾病和covid-19人类靶点基因的接近度
2.药物重定位
我们系统评估了SARS-CoV-2人类靶基因/蛋白质与6种主要类别的64种疾病的网络邻近性:自身免疫,癌症,心血管,代谢,神经和肺。
单细胞测序表明发炎性肠病的吸收性上皮细胞中的ACE2和MPRSS2升高,表明covid-19和发炎性肠病之间存在共同的病理生物学特性
哮喘患者的bulk和单细胞数据的综合分析表明,COVID-19与哮喘(包括IRAK3和ADRB2)具有中等的炎症分子特征。
