说明:此篇笔记系2016-2017年由克里克学院与康昱盛主办的蛋白质组学网络大课堂整理而成,侵删。该课程由上海易算生物科技有限公司CEO沈诚频博士所授。
主要知识点:
--质谱数据格式
--结果报告的质控
--DDA模式的工作原理
质谱数据格式
话说,蛋白质质谱从十几年前就形成了固定的数据结构和格式。现在常用的搜库格式,比如mascot的mgf,从十年前就基本固定下来。
到目前为止,质谱界的数据格式因为仪器的不同,有几个不同的大类:
Thermo公司的raw文件格式,这是目前用得最多的一种格式
AB公司的WIFF格式,
Bruker的yep/.fid
Waters的folder
Agilent的folder
Notes
MALDI MS目前应用越来越少,而且基本上不用于shotgun或者高通量研究。
这些数据格式的扩展名有一定的差别,且原始数据里包含的内容也有所不同。具体包含哪些重要的信息,稍后我们还会详细讲到。
结果报告的质控
数据分析,最终都是为了拿到一个可信的结果。所以,我们在讲具体的分析原理之前,先得来聊聊,我们做一次高通量的蛋白质定性、定量实验,以及搜库鉴定及定量分析等步骤,对结果报告有哪些质控要求。
首先,我们做完实验,在拿到下机数据的时候,大多数小伙伴们都会把数据放到各种搜库软件中,比如Mascot或者Thermo的Proteome Discoverer,导入原始数据,设定一些搜库参数,就可以得到结果了。
但是,作为一个严谨的实验方案设计来说,在分析的过程中,是需要对自己的数据有一个前期质控的,这样可以帮助大家判断数据分析结果的可靠性。所以说,基本的质控可以帮助我们对实验结果进行一个预判。
举个例子。
我们打开一个实验的下机数据,就可以预判我们的样品中是否发生了高分子塑料的PEG污染,有没有超高丰度的蛋白,或者有没有被严重的盐类污染。这些数据都可以从原始数据的可视化视图中看到。
不同的质谱软件,打开原始数据的方式不同,但这些信息都是可见的。另外,当两次实验搜索到的蛋白数量差异比较大时,也可以从TIC图来判断其原因。此外还可以判断分离的效率,以及是否出现喷雾中断等情况。
对于蛋白鉴定的结果,或者绝大多数的搜库算法,都要求对结果进行FDR控制,以及unique peptide的控制等等。如果我们要发表这些数据,绝大多数的期刊杂志也都会要求提供这些质控的信息。
那么,问题就来了,为什么要做这样的要求呢?
事实上,我们做好了质控,就能够看到一个总的鉴定的比例。比如说像常规的定量实验,用的最多的是iTRAQ。
举个例子。
假设总蛋白数只有2446个,算是比较少的,而总的谱图数是53万张,那么它的谱图鉴定率在当前条件下是32%(有些质控软件可以直接报告谱图鉴定率,比如Scaffold),我们可以判断当前的实验并没有出现重大的问题,鉴定率不高主要是因为存在高丰度蛋白,而这个后续可以进行详细的查看。
对于定量实验,不管我们使用的是SILAC,iTRAQ还是Label Free,都需要对定量结果进行准确性控制(详细内容,后续课程还会展开讲解)。一般来说,我们需要用相应的软件和统计方法来进行质控。
经过这几步的判断之后,可以得到一个初步的结果,比如说谱图数量是否和之前的结果差不多,质量精度及鉴定率如何,高丰度蛋白的存在与否,是否受污染,分离效率如何,定量是否准确,标记效率是否ok,等等,这些信息都可以得到。这样,我们最终可以得到一个准确可靠的蛋白质组学鉴定或定量结果用于后续的分析了。

那么,如何通过查看原始数据来进行初步质控呢?
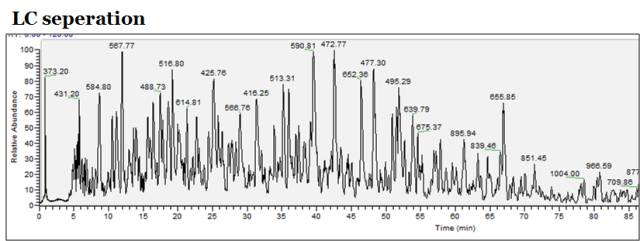
首先,我们从原始数据出发,可以看到下图(以Data-dependent-acquisiton数据依赖性扫描为例),是从色谱出来的一个LC分离得到的TIC图,其中的信号采集都是在质谱中完成的,它其实就是将色谱逐渐通过喷雾的方式进入质谱的那些信号进行逐一的扫描,然后在其中挑选高强度的谱峰进行二级碎裂。
关于LC分离,以及TIC图的详细介绍,请参考上一节课的内容:
3. 蛋白质谱的原理及使用(4)
下图就是色谱离子流图的某个瞬间。横坐标是质荷比,纵坐标是信号强度。这个瞬间进入色谱的有这样一些信号,信号强度最高的是质荷比为477.31的肽段,其他一些肽段也可以进行查看。
这是我们在打开质谱的下机数据所能看到的最直观的结果。我们需要了解的是,这只是我们所有结果的某一个瞬间,某一个scan。这一个scan是否能够反映整个结果的好坏是不确定的,所以后续我们需要进一步的展开。
对于质谱来说,在这一步会自动选择其中一个比较强的峰,比如说477,它会进行一个动态的排除,这也是Data-dependent-acquisiton的一个重要参数。就是说,在多少秒之内,这么强的一个峰如果一直反复出现的话,那么在后续的扫描过程中,我们不去再对它进行进行MS2碎裂了。
比如说如图的477.31,我们质谱仪器记录时发现前面已经对它做过二级碎裂了,那么我们就有可能选择另外一个比较弱的谱峰。比如552.80,将它进行二级碎裂。
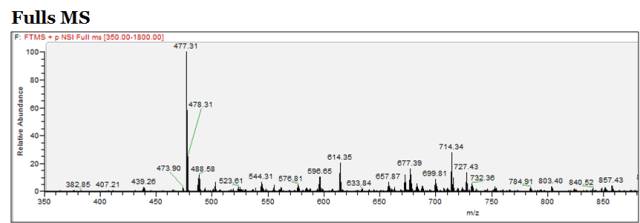
我们再来看一眼二级谱峰,如下图,就是对我们全长的进入质谱的肽段信息进行打碎,得到相应的B/Y离子,如下图,这些在后面我们会进行详细的讲解。
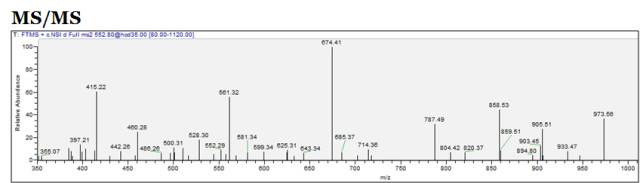
DDA模式的工作原理
下图是Thermo质谱的原理示意图(由Thermo工程师提供)。这是QE的原理图,我们先在绿色的范围内进行一次full scan的mass扫描,然后判断当前选择的离子信号强度,以及在最近的几十秒钟之内是否对其进行扫描过。
如果没有,那么在紧接着的循环过程中,我们会对之前30秒之内(假设当前的仪器速度可以达到10个MS)没有扫描过的最强的十个谱峰进行二级碎裂,那么质谱就会依次将色谱推进来的喷雾中的肽段进行依次碎裂。
这就是DDA模式基本的原理。我们的数据也是根据这样的一个过程来记录的。
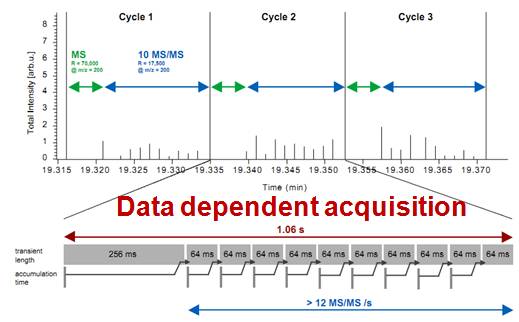
如果将刚才的扫描过程二维展开,可以得到下图,看上去跟二维凝胶电泳图很像吧?横坐标是质荷比,纵坐标是保留时间,而刚才那张图横坐标是保留时间,纵坐标是强度(LC seperation图),所以,此图没有质荷比信息。
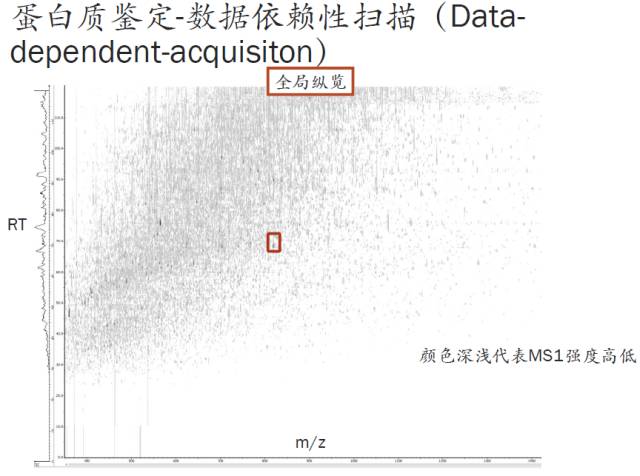
我们知道,在进入full scan的MS扫描时是有质荷比信息的。所以简单的讲,上图是将刚才的两张图的信息拼接,然后将整个下机数据所有的瞬间都进行了一个拼接,由于维度的限制,因此信号强度信息无法再展示了。
但在此图中用了颜色的深浅来表示保留时间,颜色深的就是相对信号较强的肽段。而图中的每一根小线段都代表一个肽段,小线段的长度对应着肽段的保留时间,加上横坐标质荷比的信息,因此通过这张全局纵览图,就能够看到我们这次实验分离的效果如何,有没有PEG、盐、或者其它污染,有没有喷雾中断等情况发生,这些都能在这张图中有一个大致的把握。
因此,这张图对于我们进行数据质控非常有用。不同的软件和仪器有不同的方法来提供这张图。此次举例用的图是由Peaks软件得来的。
我们可以在上图中选定自己感兴趣的部分,画一个小方框,将方框中的内容进行打开放大,就得到了下图我们存储数据的结果形式了。这是在Qual Browser里打开我们的数据看到的结果。
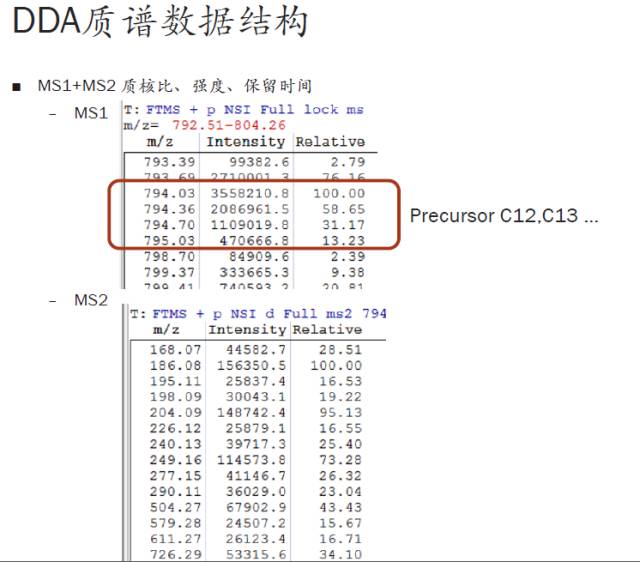
其实这就是将我们的模拟图转换成数据信号,储存在我们的Raw文件中,或者说进一步提取成MGF文件所用到的相关信息。
这里主要包含两大类信息:MS1和MS2的信息,也就是full scan mass和二级碎裂的信息。这两类信息的结构式是一模一样的,都是包含质核比、强度值,以及相对信号强度。
比如说794.03谱峰,相对信号强度是100,也就是在这张谱图中,这是最强的一个峰,信号强度是3558210.8。那么对于我们质谱的搜索来说,一级信息和二级信息都是需要用到的,其中一级信息是首要的,也就是图中MS1部分,是后续搜库的关键信息。而二级谱图的强度信息一般用于定量,也就是说如果不是做SILAC或者非标记定量,这些信息不是最重要的。
另外,第一栏的信息准确性也是非常重要的。比如图上红框内,我们可以得到的信息是,794.03和794.36强度大约差了1.5倍,后面的峰强度差了大约2倍,再看下红框内四个数据的质荷比相差并不大,我们的质谱仪器因此会判断这四个峰非常符合一个肽段的同位素分布(肽段同位素分段的性状,后续将会讲解)。
回到此图,794.03应该是一个肽段,后面三个数据是同一个肽段,这就是我们进行precursor识别的原理。有些时候质谱会识别错误,认为红框上一行的793.69更可能是同位素,这个就需要我们自己进行校正。
质谱在搜集信号的时候,会告诉我们794.03是一个母离子或者说是肽段的谱峰,因此在后续进行MS2碎裂的时候,会挑选这样一个谱峰,以及在质谱中我们会设定相应的窗口去打碎它。因为仅仅设定一个非常小的窗口,可能信号不够。我们会设计比如正负1.5个道尔顿的窗口,把这些信号全部采集进去进行二级碎裂得到二级信号。
现在高分辨质谱中,二级信号也会包含同位素信息,因此数据分析软件需要对这些信息进行有效的处理。
大家可以看到,这样一个例子中,软件记录的是794.03,但实际我们可以通过肉眼观察,793.69跟794.03就只相差0.33~0.34,也是一个三电荷同位素的差值(1除以0.33是3,这就是质荷比中的Z的计算原理)。两者分别的强度271万和355万差别也不是非常大,我们会判断出793.69更可能是零同位素峰(如何判断后面会再讲解)。
我们进行后续数据提取和采集的时候,也就是用了这样的信息来进行分析。我们记录的一级质谱数据,以及二级质谱对应的列表,其中最重要的是m/z和intensity,在一级质谱数据中,强度并不用于蛋白鉴定的打分,但二级质谱数据中的强度值却会被用于打分。
下篇将聊聊同位素的问题,以及如何解读原始谱图包含的信息。