最近在做样品比较多的转录组分析项目,有部分样品bam文件极大。所以导致用featurecounts去进行基因的count过程出奇的慢!!!,目前已经运行两天有余,还在LEVELMERGE和MERGE阶段“墨迹”,真他妈墨迹,我也搞不懂LEVELMERGE和MERGE这两个过程有啥区别,为啥不同,我眼中怀疑前一个阶段可以多线程,走到这里就单线程了####我猜的,因为看不太懂R脚本,屌大的大神可以给我讲解一下区别和过程####(传说中的featurecounts优于HTseq等一众定量软件,快如狗那???)。
迷茫之中,查阅网站,发现两个快如闪电,Aligment-free的转录组分析软件。这不是我完全不懂生信时,预期的转录组分析的样子吗。给你想要的基因数据集,给我直接用reads比,给我表达量,干净利落,爱了。
声明警告:
总结的超级好了,我就直接转载了,学习记录用,不做商业用途。
使用salmon和kallisto进行RNA-seq定量(包括文献阶段,步骤,及个人使用体会):http://blog.sciencenet.cn/blog-1094241-1133526.html
Alignment-free的转录本比对工具-Salmon: https://www.bioinfo-scrounger.com/archives/411/
RNA-seq从入门到自闭(Kallisto和Salmon)https://www.jianshu.com/p/86a409a65ed3
salmon要优于kallisto和express软件
介绍下其定量流程,与kallisto进行一个定量结果的比较,以及看一看基因拷贝数变化时TPM如何变化。
首选要对转录本进行index
salmon index --keepDuplicates -t IWGSC_v1.1_HC_LC_20170706_transcripts.fasta -i IWGSC_v1.1_HC_LC
下载测试数据,该测试数据来自中国春的叶片。
axel -a -n 12 ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR947/SRR947005/SRR947005_1.fastq.gz && axel -a -n 12 ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR947/SRR947005/SRR947005_2.fastq.gz
进行定量
salmon quant -i IWGSC_v1.1_HC_LC -l A -1 SRR947005_1.fastq.gz -2 SRR947005_2.fastq.gz -p 8 -o SRR947005_quant
-i 指要使用的转录组index 这里是我们第一步生成的IWGSCv1.1版本的转录组index;
-l 这里需要指定测序模式,这里-A是指自动判定。
-1 -2 分别要输入左端测序数据和右端测序数据
-p 则是要指定运行程序的cpu线程数
上述定量命令使用测序的原始reads进行的,这里还有另外一种基于比对的方式,即输入文件是BAM或SAM文件。
输出的结果文件如下所示,其中quant.sf里有每个基因的表达量。

kallisto定量过程
首先也是要对转录组进行index
kallisto index -i IWGSC_v1.1_HC_LC_kallisto IWGSC_v1.1_HC_LC_20170706_transcripts.fasta
其次是定量过程。
kallisto quant -i IWGSC_v1.1_HC_LC_kallisto -t 10 -o SRR947005_kallisto SRR947005_1.fastq.gz SRR947005_2.fastq.gz
同样的结果文件如下图所示,其中abundance.tsv里有每个基因的表达量。
在同一个项目中,使用的软件版本要一致。至于哪一个salmon和kallisto哪个更好,我个人可能更偏向salmon,但kallisto毕竟是science使用的,所以做小麦的话,kallisto应该是公认的。kallisto算出来的差异基因数量要多于salmon。
但同时也要注意IWGSC注释的转录本的正确率。特别是对于低可信度的基因,基因结构不正确,做出来的TPM可能也不对。而且IWGSC注释的转录本其3’和5’的非翻译区有不少缺失。转录组里还可以组装出新的转录本或者新的可变剪切。
目前IWGSC给出的转录本是26万左右,而其中高可信的基因只有10万。我们进行差异表达分析时使用的是26个转录本,但只有那10万个高可信基因的功能注释较为可信。所以后续的功能富集分析,就要慎重。差异表达可以包括低可信的基因,但功能富集分析时可以不包括在里面,特别是不能用来充当总数,science上的那篇文章也正是这样做的。另外,功能富集分析时使用的背景基因应该是在某条件下表达的基因集,而不是总基因集。比如,总共有10个基因,其中只有5个基因表达,而这5个表达的基因里有2个差异表达,此时使用的总数是5而不是10。当然,如果差异基因里包括低可信的基因,那表达的总基因里也可以包括低可信度的基因。不过,我还是建议使用表达的高可信度的基因作为整体。
精品RNA分析软件| kallisto,卡丽丝多 (https://www.sohu.com/a/204272047_99971433)
我们说过现在比较流行的RNA分析套路有hisat2+stringtie、tophat+cuffflinks或者star+htseq等,但是这些都是基于首先将read回帖到基因组上再进行表达定量计算的,换句话说也有一些软件并不需要将read进行回帖,可以直接通过分析read属于哪一个基因来计算基因的表达量,如本篇要讲的kallisto就是这类软件的一个代表。
该软件2015年被发表在NATURE BIOTECHNOLOGY上,Title:《 Near-optimal probabilistic RNA-seq quantification》,被引用量超过150,下面老牛以自己的理解为大家稍微解读下这篇文章。
首先作者吐槽了一下TopHat2这个软件的效率问题,其实TopHat2本身对多核的优化就不好,作者用20
核运行TopHat2有点无疑更加放大TopHat2的劣势,而且作者团队自身开发的kallisto index这一步也只能单核运行,而到了quant这一步才支持多线程。Cufflinks这个软件也是由于效率很低被作者提了。针对效率低的这个问题虽然可以通过streaming algorithms 或者 naive counting of reads 等方法提高速度,但同时也牺牲了精确度。为了避开alignment这一步,最近的一个研究建议可以提取read的k-mers并且同时使用k-mers的哈希表。但是由于read的k-mers可以比对到多个转录本上,所以就造成了其丢失了一定的精确度。
虽然直接使用k-mers相关方法并不好,但是hash-based方法确实可以提高运行速度,作者因此试图以此为切入点开发新的算法。作者发现,如果想要精确地定量转录本并不需要准确的知道reads被定位到哪里,只需要知道该reads被哪个转录本拥有就可以了。基于此,作者开发了一个pseudoalignment的新算法。新算法的测试结果非常好,下面就挑几个主要的方面进行讲解。
首先是处理模拟数据,与bowtie2相比,kallisto和bowtie2都找出了同一个测试样本的70.7%的转录本,但是具体到单个read平均比对的转录本数量上,kallisto的4.96要低于bowtie2的8.02。在真实样本处理中,两者共同比对到的read达到66.22%,但是kallisto的单个read比对上的4.86个转录本依旧要少于bowtie2的8.94个转录本。
作者使用模拟数据基于count进行了精确度上的评测,kallisto有着很好的表现,远超出TopHat2+Cufflinks、HISAT+Cufflinks和Sailfish等软件。另外,作者用有qPCR数据的SEQC data数据进行了测试,kallisto的表现和其他软件的结果类似。
说完了最基本的组装精确度评测,下面作者就开始讲kallisto最大的优势了:超快的运行速度。对一个3千万read的模拟数据只需要单核7.5分钟就能完成,在众多的软件中是最优秀的。除了超快的运行速度,kallisto对内存的使用效率也比较高,单个模拟样本只需要3.2Gb的内存,降低了RNA分析的硬件标准。
kallisto的参数也非常少,下面就为大家稍微讲解一下kallisto的用法。
首先我们找到kallisto的官方下载地址:
https://pachterlab.github.io/kallisto/download,下载最新的v0.43.1 Linux版本,tar -zxvf 解压文件。
1.创建索引
kallisto index [arguments] fastq
参数: -i生成的索引文件的文件名
-kk-mer的大小,默认为31,最大也是31
2.计算定量
kallisto quant [arguments] fastq
参数: -i输入的索引文件
-o输出目录
-t使用的线程数默认为1
--single针对单端序列
-l 估算的平均片段长度大小,单端数据用
-s 估算的片段长度标准误差,单端数据用
示例,双端测序数据:kallisto quant -i index -o output pairA_1.fastq pairA_2.fastq
单端测序数据:kallisto quant -i index -o output --single
-l 200 -s 20 file1.fastq
kallisto quant的输出文件有三种类型:
abundances.h5可以被R包软件sleuth读取的文件,包含了丰度估计、转录本长度等等信息。
abundances.tsv包含丰度估计的纯文本文档。
run_info.json包含软件运行信息。
接下来的操作就需要在另外一个R包软件sleuth上进行了,后续的推文里会讲到这个软件的使用方法。
使用salmon和sleuth进行小麦RNA-seq差异表达分析(https://mp.weixin.qq.com/s/ONRTfJUvmGQjIw2EqApOew)
好了,我们言归正传。上次我们在“评估salmon和kallisto在小麦RNA-seq定量中的异同”中论证了salmon能够充分区分小麦的同源基因,哪怕只有一个SNP也能够有效区分。同时我们也发现,早期版本的kallisto有重大bug,要尽快升级到最新版本。以前使用kallisto进行定量时,有专门的软件sleuth进行差异表达分析。那么sleuth是何方神圣?2017年sleuth被发表在nature methods。谷歌显示至今大约被引用116次。自然要比我们常用的DESeq2,edgeR等软件的准确率要高。

image-20180926115001362
下面我们就重点谈一谈如何使用这两个软件进行差异基因的表达分析。在实际使用过程中发现里边有很多坑。
第一部分 参考转录组
参考转录组也即中国春参考基因组上注释出来的基因序列。参考转录组当然越全面越准确才好。变好总得需要一个过程。目前1.0版本的数据有如下几个不足:首先,基因的3’和5’端比较短,没有到头;第二是,很多基因具有多个转录本,1.0给的比较少;第三,遗漏了不少转录本。
具体到我们手里的数据,我们可以采用以下做法。也即先找出自己数据中的新转录本或新基因,然后补充进参考转录组进行下一步的差异表达分析。这样有利于发现数据中独特的转录本或基因。当然了,如果只用中国春1.0的参考转录组,对最终的结果影响也不大,例如,功能富集分析的结果等。这些比参考转录组多出来的基因,往往是功能未知基因,没有保守结构域等,可以根据表达等特征筛选比较有趣的进行具体的研究。
下面,我们谈一谈如何获取相对中国春1.0的新转录本和新基因。
1 使用STAR将reads映射至中国春参考基因组。这一步也可以使用其他mapping软件,如hisat2, bowtie2, bbmap等。因为处理的样本较多,我这里使用python写了一个脚本处理。熟悉shell的也可以写shell脚本。
#!/usr/bin/env python
# -*- coding: utf-8 -*-
__author__ ='wheatomics'
importsubprocess
withopen('input.txt','r')asf:
forlineinf:
line = line.strip().split()
fq1, fq2 = line
printfq1, fq2
proc = subprocess.Popen(
['STAR','--twopassMode','Basic','--genomeDir','/data/genome_whole/',
'--runThreadN','30','--limitSjdbInsertNsj','5000000','--outSAMtype','BAM','SortedByCoordinate','--twopass1readsN','-1','--readFilesCommand','zcat',
'--sjdbOverhang','100','--outSAMattributes','All','--outSAMmapqUnique','60','--outFilterMismatchNmax','10','--readFilesIn', fq1, fq2,
'--outSAMattrRGline','ID:'+ fq1.split('/')[-1].split('.')[0],'SM:'+ fq1.split('/')[-1].split('.')[0],'PL:ILLUMINA','--outFileNamePrefix',fq1.split('/')[-1].split('.')[0]], shell=False)
proc.wait()
# 注意这个参数 --outSAMmapqUnique 60,这是为了后面使用GATK call variants. 如果没有这个需求,可以不用设置,但是如果call variants又只能将一整条染色体分成两部分。所以不能两全其美。
2 筛选bam文件。这里将bam文件里unmapped的reads去掉;因为,我们的样品来自中国春的叶片,所以大于一个mismatch的reads也去掉;去掉mapping长度小于80的reads;只保留proper pair的reads。
for i in {CS_CT_1_1,CS_CT_2_1,CS_CT_3_1,ABA1h_1_1,ABA1h_2_1,ABA1h_3_1,ABA12h_1_1,ABA12h_2_1,ABA12h_3_1,ABA24h_1_1,ABA24h_2_1,ABA24h_3_1};do bamutils filter $i.sorted.bam $i.filtered.sorted.bam -minlen 80 -mapped -properpair -mismatch 1;done
顺带交代下样本的背景。这里是对中国春叶片外施ABA后,分别在1h,12h,24h取样。每个时间点3个重复。这是我现编的。
3 组装转录本。输入文件是上面筛选过的bam文件。软件使用scallop。scallop是去年发表在nature biotechnology上的一款有参转录本组装软件。

image-20180927145240879
据作者的评测要比过往的软件好。但这并不意味着使用二代测序reads组装出来的转录本就是无比正确的,这一点要铭记,特别是当要针对具体的某条转录本进行研究时。
On 10 human RNA-seq samples, Scallop produces 34.5% and 36.3% more correct multi-exon transcripts than StringTie and TransComb, and respectively identifies 67.5% and 52.3% more lowly expressed transcripts. Scallop achieves higher sensitivity and precision than previous approaches over a wide range of coverage thresholds.
for i in {CS_CT_1_1,CS_CT_2_1,CS_CT_3_1,ABA1h_1_1,ABA1h_2_1,ABA1h_3_1,ABA12h_1_1,ABA12h_2_1,ABA12h_3_1,ABA24h_1_1,ABA24h_2_1,ABA24h_3_1}; do scallop -i ${i}.filtered.sorted.bam -o ${i}.gtf; done
上述命令运行完之后会产生12个gtf文件。下面要做的就是合并这12个gtf文件中的转录本。这里使用 stringtie merge命令。
4 合并转录本。其中iwgsc_refseqv1.0_HCandLC.gtf来自官方的注释文件。mergelist.txt包含上述12个gtf文件的名字,每行一个,共12行。
stringtie --merge -p 8 -G /data/IWGSCv1.0/iwgsc_refseqv1.0_HCandLC.gtf -o meng_merged.gtf ./mergelist.txt
5 与官方注释信息比较,找出新增的转录本或基因。
#比较
gffcompare -o gffall -r /data/IWGSCv1.0/iwgsc_refseqv1.0_HCandLC.gtf all_merged.gtf
# gffcompare 命令来自 https://ccb.jhu.edu/software/stringtie/gffcompare.shtml
# 分离新转录本
gtfcuff puniq gffall.all_merged.gtf.tmap all_merged.gtf /data/IWGSCv1.0_RefSeq_Annotations/iwgsc_refseqv1.0_HCandLC.gtf all_merged_unique.gtf
# gtfcuff 命令来自 https://github.com/Kingsford-Group/rnaseqtools
6 获取新增转录本的序列。
gffread all_merged_unique.gtf -g data/IWGSCv1.0/genome.fasta -w all_merged_unique.fasta
# gffread 命令来自 http://ccb.jhu.edu/software/stringtie/gff.shtml
7 新转录本和中国春参考转录本合并。
cat data/IWGSCv1.0/iwgsc_refseqv1.0_HCandLC.fasta all_merged_unique.fasta > all_merged.fasta
至此新转录本获取完毕。下面选取2个与参考转录本比较下。
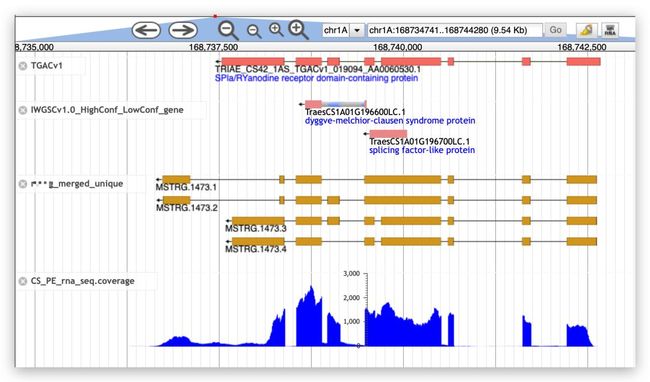
上图显示了与参考基因组不同的转录本

新基因,这里也有TGAC的转录本支持
第二部分 基因定量
基因的定量使用salmon这个软件。使用的转录本就是上述我们合并而成的。
# index
salmon index -p 4 -t all_merged.fasta -i all_merged_salmon_index
# 定量
for i in {CS_CT_1_1,CS_CT_2_1,CS_CT_3_1,ABA1h_1_1,ABA1h_2_1,ABA1h_3_1,ABA12h_1_1,ABA12h_2_1,ABA12h_3_1,ABA24h_1_1,ABA24h_2_1,ABA24h_3_1};do salmon quant -i Uall_merged_salmon_index -l A -1 ../clean/${i}_1_clean.fq.gz -2 ../clean/${i}_2_clean.fq.gz -p 8 --numBootstraps 100 -o ${i}_quant;done
# 参数 --numBootstraps 100 一定要放上
# 将生成的以“_quant”结尾的文件夹统一放到salmon_result文件夹下面.
mkdir salmon_result
mv *_quant salmon_result/
至此,定量部分完成
第三部分 差异表达分析
这里使用sleuth。sleuth本来是为kallisto量身定制的,如果salmon想使用sleuth进行差异表达分析,则需要一个R包(wasabi)进行转换。
1 安装wasabi
# 地址 https://github.com/COMBINE-lab/wasabi
source("http://bioconductor.org/biocLite.R")
biocLite("devtools")# only if devtools not yet installed
biocLite("COMBINE-lab/wasabi")
或者使用 conda install r-wasabi安装
2 结果转换
library(wasabi)
sfdirs <- file.path("salmon_result", c("CS_CT_1_quant","CS_CT_2_quant","CS_CT_3_quant","ABA1h_1_quant","ABA1h_2_quant","ABA1h_3_quant","ABA12h_1_quant","ABA12h_2_quant","ABA12h_3_quant","ABA24h_1_quant","ABA24h_2_quant","ABA24h_3_quant"))
prepare_fish_for_sleuth(sfdirs)
3 使用sleuth进行差异表达分析
# 这里需要一个分组信息文件“sample_infor.txt”,如下格式,两列。注意,除了表头,第一列的顺序要按字母顺序排列
sample condition
ABA1h_1 ABA1h
ABA1h_2 ABA1h
ABA1h_3 ABA1h
ABA12h_1 ABA12h
ABA12h_2 ABA12h
ABA12h_3 ABA12h
ABA24h_1 ABA24h
ABA24h_2 ABA24h
ABA24h_3 ABA24h
CS_CT_1 CS_CT
CS_CT_2 CS_CT
CS_CT_3 CS_CT
#为了获取基因水平的差异,需要一个转录本和基因对应的文件“transcript_gene_relation.txt”。
#文件包括两列,第一列是转录本名字,第二列是基因名字。
transcript_id gene_id
TraesCS1A01G425600.1 TraesCS1A01G425600
TraesCS1A01G425700.1 TraesCS1A01G425700
TraesCS1A01G425800.1 TraesCS1A01G425800
TraesCS1A01G602400LC.1 TraesCS1A01G602400LC
TraesCS1A01G602500LC.1 TraesCS1A01G602500LC
我们这里是按时间点取样,可以按照时序分析,不必进行两两比较。
library(sleuth)#加载包
library(splines)#加载包
sample_id <- dir(file.path(".","salmon_result"))#加载salmon_result下的文件夹
sample_id#显示有多少样品
kal_dirs <- file.path(".","salmon_result", sample_id)
kal_dirs
s2c <- read.table(file.path(".","sample_infor.txt"), header =TRUE, stringsAsFactors=FALSE)
s2c <- dplyr::select(s2c, sample = sample, condition)
s2c <- dplyr::mutate(s2c, path = kal_dirs)
s2c#注意查看是否一一对应,这个地方一不小心会出现对应错误。也即为啥要求sample_infor.txt里要按第一列排序的原因。
design <- model.matrix(~ -1+factor(c(1,1,1,2,2,2,3,3,3,4,4,4)))#此处的design我不是十分确定是否准确?
colnames(design) <- c("CS_CT","ABA1h","ABA12h","ABA24h")
design
t2g <- read.table(file.path(".","transcript_gene_relation.txt"), header =TRUE)
t2g <- dplyr::rename(t2g,target_id = transcript_id, gene_id = gene_id)
so <- sleuth_prep(s2c, full_model = design, num_cores =1,target_mapping = t2g,aggregation_column ='gene_id', extra_bootstrap_summary=TRUE,read_bootstrap_tpm=TRUE, gene_mode =TRUE)
#参数 num_cores = 1 一定要设为1,不然会报错。
#如果读取出现错误,可能是因为R包rhdf5的问题,即wasabi和sleuth依赖的rhdf5不同。我这里采取的策略就是分别安装在不同环境里了。或使用conda install --channel bioconda r-sleuth。将wasabi安装在wasabi环境里,conda create -n wasabi r-wasabi。这个环境与sleuth的环境不同
so <- sleuth_fit(so)
so <- sleuth_fit(so, formula = ~1, fit_name ="reduced")
so_lrt <- sleuth_lrt(so,"reduced","full")
models(so_lrt)
sleuth_table <- sleuth_results(so_lrt,'reduced:full','lrt', show_all =FALSE)
sleuth_significant <- dplyr::filter(sleuth_table, qval <=0.05)
head(sleuth_significant)
write.csv(sleuth_table,"CS_ABA_sleuth_gene_level.csv",row.names=TRUE,quote=TRUE)
write.csv(sleuth_significant,"CS_ABA_sleuth_significant_gene_level.csv",row.names=TRUE,quote=TRUE)
sleuth_matrix <- sleuth_to_matrix(so_lrt,'obs_norm','tpm')
head(sleuth_matrix)
write.csv(sleuth_matrix,"CS_ABA_sleuth_tpm_norm_gene_level.csv",row.names=TRUE,quote=TRUE)
我们也可以进行两两比较
#首先要明确的是,是那些样本之间进行两两比较。此处是:CS_CT vs ABA1h, CS_CT vs ABA12h, CS_CT vs ABA24h, ABA1h vs ABA12h,ABA1h vs ABA24h, ABA12h vs ABA24h。共六组
library(sleuth)#加载包
library(stringr)#加载包
sample_id <- dir(file.path(".","salmon_result"))#加载salmon_result下的文件夹
kal_dirs <- file.path(".","salmon_result", sample_id)
s2c <- read.table(file.path(".","sample_infor.txt"), header =TRUE, stringsAsFactors=FALSE)
s2c <- dplyr::select(s2c, sample = sample, condition)
s2c <- dplyr::mutate(s2c, path = kal_dirs)
s2c#注意查看是否一一对应,这个地方一不小心会出现对应错误。也即为啥要求sample_infor.txt里要按第一列排序的原因。
t2g <- read.table(file.path(".","transcript_gene_relation.txt"), header =TRUE)
t2g <- dplyr::rename(t2g,target_id = transcript_id, gene_id = gene_id)
#下面的语句进入两两比较
a <- list(c("CS_CT","ABA1h"), c("CS_CT","ABA12h"), c("CS_CT","ABA24h"), c("ABA1h","ABA12h"),c("ABA1h","ABA24h"), c("ABA12h","ABA24h"))
for(xxxina) {
s2b <- dplyr::filter(s2c, condition == xxx[1] | condition == xxx[2])
so <- sleuth_prep(s2b, ~ condition, num_cores =1,target_mapping = t2g,aggregation_column ='gene_id', extra_bootstrap_summary=TRUE,read_bootstrap_tpm=TRUE,gene_mode =TRUE)
so <- sleuth_fit(so)
so <- sleuth_fit(so, formula = ~1, fit_name ="reduced")
so_lrt <- sleuth_lrt(so,"reduced","full")
models(so_lrt)
sleuth_table <- sleuth_results(so_lrt,'reduced:full','lrt', show_all =FALSE)
sleuth_significant <- dplyr::filter(sleuth_table, qval <=0.05)
head(sleuth_significant)
write.csv(sleuth_table,str_c(xxx[1],"vs", xxx[2],"_sleuth_gene_level.csv"),row.names=TRUE,quote=TRUE)
write.csv(sleuth_significant,str_c(xxx[1],"vs", xxx[2],"_sleuth_significant_gene_level.csv"),row.names=TRUE,quote=TRUE)
sleuth_matrix <- sleuth_to_matrix(so_lrt,'obs_norm','tpm')
head(sleuth_matrix)
write.csv(sleuth_matrix, str_c(xxx[1],"vs", xxx[2],"_sleuth_tpm_norm_gene_level.csv"),row.names=TRUE,quote=TRUE)
}
sleuth还提供了一些画图函数,有兴趣的可以自行尝试。
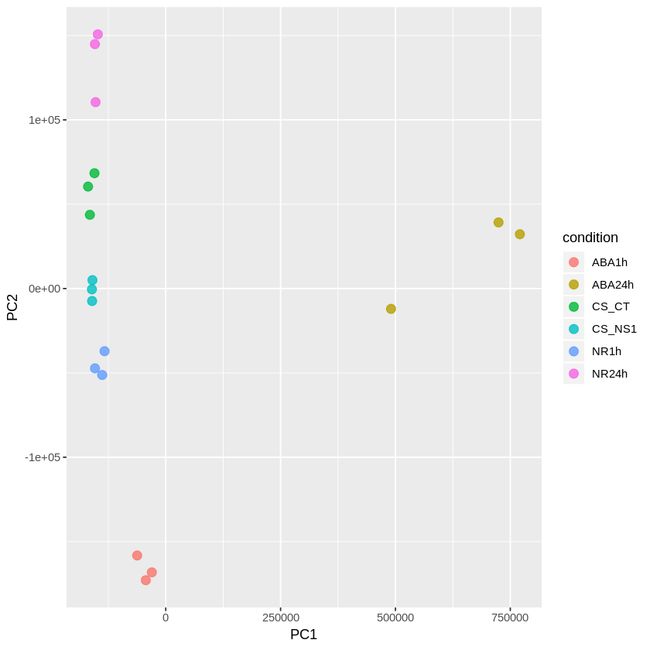
PCA
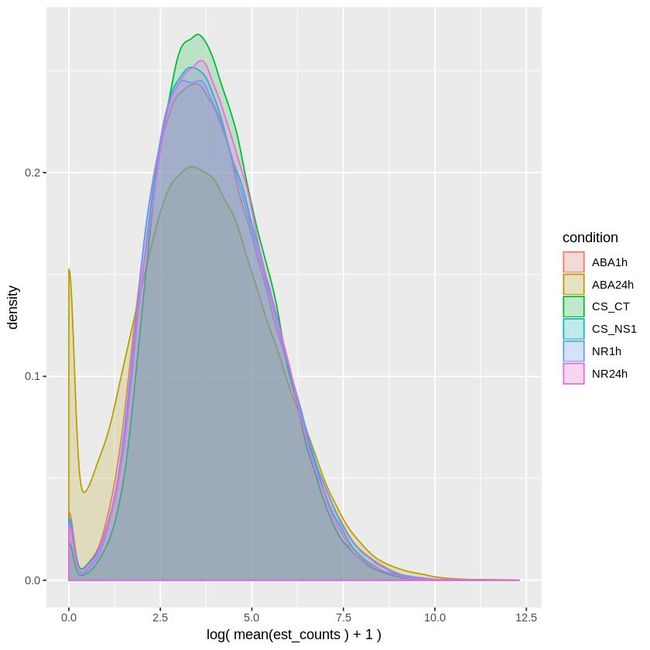
samples_density
sleuth结果里只提供了q value 来筛选差异表达基因,没有倍数变化的结果。这里可以使用excel自行进行计算。
计算 Fold change(倍数变化),计算方法如下:
E = mean (group1) B=mean (group2)
FC= (E-B) / min (E, B)
基因 A 和基因 B 的平均值之差与两者中较小的比值。选择 2-3 倍的基因作为差异表达基因即可。可以根据具体数据,斟酌使用。
提供一个小麦里的文献作为参考吧。今年Cristobal Uauy在BMC Plant Biology上以通讯作者发表的文章,题目是“Ubiquitin-related genes are differentially expressed in isogenic lines contrasting for pericarp cell size and grain weight in hexaploid wheat”。

BMC Plant Biology
