hello,大家好,今天我们来分享一个实用的方法,SIGMA,这个方法是用来做什么的呢??就是说我们对一个样本进行初次聚类之后,很多时候我们需要对一个cluster再分群看看,是否一个cluster内部也具有异质性,从而挖掘有意义的新的细胞类型,那如何判断一个cluster是否值得再分群分析呢??这个软件可以辅助我们来分析一下,文章在A clusterability measure for single-cell transcriptomics reveals phenotypic subpopulations,我们简单回顾一下文献,重点看看实例代码。
文章最重要的结论就是对每个cluster计算一个SIGMA值,值越接近于1,说明一个cluster越包含有意义的subcluster,很有必要进行cluster的再分群分析。
Abstract
The ability to discover new cell populations by unsupervised clustering of single-cell transcriptomics data has revolutionized biology(这个应该是共识了). Currently, there is no principled way to decide, whether a cluster of cells contains meaningful subpopulations that should be further resolved(当前,没有主要的方法来确定一群细胞是否包含有意义的亚群). Here we present SIGMA, a clusterability measure derived from random matrix theory(随机矩阵理论), that can be used to identify cell clusters with non-random sub-structure, testably leading to the discovery of previously overlooked phenotypes.
Main,这个地方我们总结一下
1、All existing clustering algorithms have adjustable parameters, which have to be chosen carefully to reveal the true biological structure of the data
2、If the data is over-clustered, many clusters are driven purely by technical noise and do not reflect distinct biological states.(不能分群过细)。
3、If the data is under-clustered, subtly distinct phenotypes might be grouped with others and will thus be overlooked.(分群数过少容易掩盖一部分异质性)。
4、现有的评估聚类质量的工具(例如广泛使用的轮廓系数)无法揭示聚类内的可变性是否是由于亚群或随机噪声的存在。
5、为了缓解这个问题,作者开发了软件SIGMA,We consider clusterability to be the theoretically achievable agreement with the unknown ground truth clustering, for a given signal-to-noise ratio.
6、Importantly, our measure can estimate the level of achievable agreement without knowledge of the ground truth
7、High clusterability (indicated by SIGMA close to 1) means that multiple phenotypic subpopulations are present and clustering algorithms should be able to distinguish them.
8、Low clusterability (indicated by SIGMA close to 0) means that the noise is too strong for even the best possible clustering algorithm to find any clusters accurately.(内部几乎没有异质性了)。
9、If SIGMA equals 0, the observed variability within a cluster is consistent with random noise.(这个时候不需要进行再分群分析)。
为了得出SIGMA值,我们将未观察到的实际基因表达谱(信号矩阵)视为对随机噪声矩阵的扰动 ,如下图:
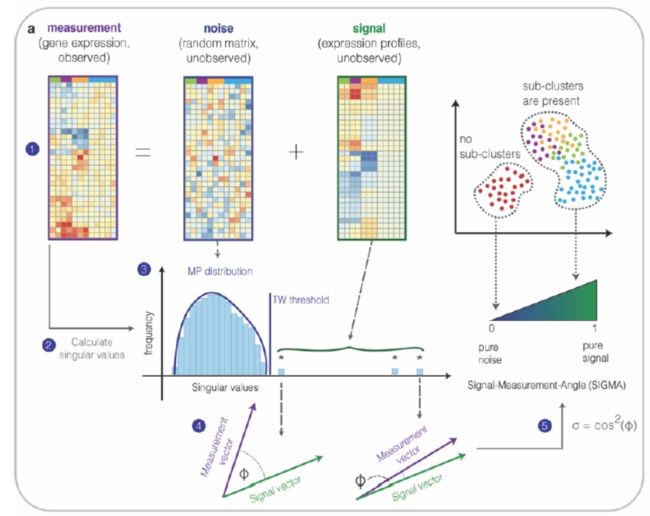
Our point of view allowed us to leverage well-established results from random matrix theory and perturbation theory。(随机矩阵理论和扰动理论)。
- first calculate the singular value(奇异值) distribution of the measured expression matrix。
if the data is preprocessed appropriately,如下图:
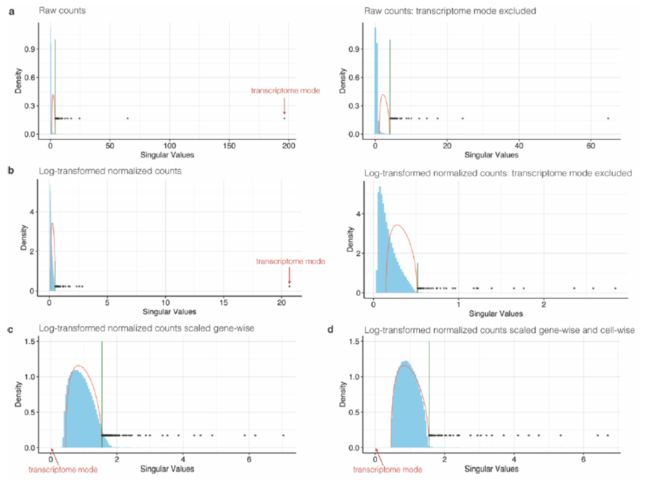
the bulk of this distribution is described by the Marchenko-Pastur (MP) distribution(随机高维矩阵), which corresponds to the random component of the measurement. The singular values outside of the MP distribution and above the Tracy Widom (TW) threshold correspond to the signal,这个地方有点难理解,因为涉及到很深的统计学知识,首先我们需要知道随机高位矩阵计算出来的奇异值符合MP分布,如果我们得到的矩阵计算出来的奇异值在这个分布以外,说明并非随机造成,而是真正的信号,不过我们简单理解一下就是高出MP分布就是我们需要的signal(当然,有一定的阈值)。接下来就是计算SIGMA值。
这个地方要注意一下:Using just these singular values and the dimensions of the measurement matrix, we can calculate the angles between the singular vectors of the measured expression matrix and those of the (unobserved) signal matrix(实际得到的矩阵和信号矩阵两个奇异值向量的角度,不知道大家还知不知道两个向量怎么求夹角,不会的翻一翻大学线性代数的书吧,作者也需要充充电了)。SIGMA is the squared cosine of the smallest angle。(SIGMA值的由来)。
Data sets with higher signal-to-noise ratios have more easily separable clusters and larger singular values outside of the MP distribution

By definition, that results in higher values of SIGMA

依据这个理论,我们来看看这个例子

图上可知,再结合之前的介绍,SIGMA值越大,越包含有意义的subcluster。

结论也是一样的。
我们重点看看示例代码
加载包
library(SIGMA)
library(ggplot2)
library(Seurat)
The authors who have anlyzed this data already normalized the data set with the R package “scran” and determined clusters by hierachical clustering. In total they have found 22 clusters.
data("force_gr_kidney")
data("sce_kidney")
paga.coord$Group <- sce_kidney$cell.type
ggplot(paga.coord, aes(x = V1, y = V2, colour = Group)) +
geom_point(shape = 16)

With SIGMA, we are now able to assess the variability for each cluster and see if possible sub-clusters can be found. First, we load the preprocessed SingleCellObject of the kidney data.
#Load kidney data from package
#Extract scran normalized counts and log-transform
expr.norm.log <- as.matrix(log(assay(sce_kidney, "scran")+1))
#Change the name of the rows to readable gene names
rownames(expr.norm.log) <- as.character(rowData(sce_kidney)$HUGO)
rownames(sce_kidney) <- as.character(rowData(sce_kidney)$HUGO)
In the next step we would like to exclude certain variances from appearing in the measure. For example, in this fetal kidney data set, several factors would not be of interest to cluster on: cell cycle related variances, ribosomal and mitochondrial gene expression. As, well as stress related genes, which arise during dissociation. Cycling genes, we determine here with the Seurat package, so for that we first need to create a Seurat object and normalize it. Another important factor is technical variability, for example the varying number of transcripts. It’s important to also include that in the data frame.
#Creating Seurat object
cnts <- counts(sce_kidney)
colnames(cnts) <- 1:ncol(cnts)
rownames(cnts) <- as.character(rowData(sce_kidney)$HUGO)
fetalkidney <- CreateSeuratObject(cnts)
#> Warning: Non-unique features (rownames) present in the input matrix, making unique
#> Warning: Feature names cannot have underscores ('_'), replacing with dashes ('-')
fetalkidney <- NormalizeData(fetalkidney)
#Cell cycle analysis
s.genes <- cc.genes$s.genes
g2m.genes <- cc.genes$g2m.genes
fetalkidney <- CellCycleScoring(fetalkidney, s.features = s.genes, g2m.features = g2m.genes, set.ident = TRUE)
#> Warning: The following features are not present in the object: MLF1IP, not searching for symbol synonyms
#Determining the expression of MT-genes, Rb-genes and stress genes:
data("ribosomal_genes")
data("stress_genes")
rb <- rownames(fetalkidney) %in% rb.genes
stress.genes <- intersect(stress.genes, rownames(expr.norm.log))
#Creating the final data frame with all the factors to be excluded from considering while calculating the clusterability measure:
exclude <- data.frame(clsm = log(colSums(cnts) + 1), cellcycle = fetalkidney$G2M.Score,
mt = colMeans(expr.norm.log[grep("^MT-", rownames(expr.norm.log)),]),
ribosomal = colMeans(expr.norm.log[rb,]), stress = colMeans(expr.norm.log[stress.genes,]))
Now we are ready to apply the main function to determine clusterability:
out_kidney <- sigma_funct(expr.norm.log, clusters = sce_kidney$cell.type, exclude = exclude)
We can have a look at the main output of this function. For each cluster, the corresponding clusterability measure is shown.
#Evaluate the output of the measure
#plot all values for sigma
plot_sigma(out_kidney)

值越大,越包含有意义的subcluster。
If you would like to go into more detail, then you can have a look at all sigmas and g-sigmas that are available per cluster.
#Plot all values for sigma and g_sigma
plot_all_sigmas(out_kidney)

plot_all_g_sigmas(out_kidney)

Data sets with higher signal-to-noise ratios are characterized by higher values of G-SIGMA。(这个地方文中提到,which indicates a more accurate estimation of differential gene expression after sub-clustering.Our approach thus not only identifies relevant sub-structure in a cell cluster but can also reveal the genes responsible for it. This is not a direct replacement for differential expression tests, but 106 a way to understand the variability within the cell-singular vectors.)
If you are interested in the values of all sigmas, g-sigmas and singular values of the signal matrix, then this information can be obtained with the help of this function.
#obtain the values for sigma and additional information
get_info(out_kidney, "UBCD")
#> sigma g_sigma theta r2vals singular_value celltype
#> 16 0.9718702 0.7595932 1.8030720 0.4468447 1 UBCD
#> 17 0.9613534 0.7073134 1.5854881 0.1810414 2 UBCD
#> 18 0.8545601 0.4459294 0.9704636 0.4134855 3 UBCD
#> 19 0.8649745 0.4617228 0.9958318 0.1402408 4 UBCD
#> 20 0.8749372 0.4779268 1.0228843 0.1069279 5 UBCD
#> 21 0.0000000 0.0000000 0.5170606 0.4340084 6 UBCD
#> 22 0.0000000 0.0000000 0.5170606 0.2978763 7 UBCD
#> 23 0.0000000 0.0000000 0.5170606 0.2157584 8 UBCD
Now, to determine if the clustrs with a high clusterability measure have variances that are meaningful for you to sub-cluster, have a look at the variance driving genes, which will tell you which genes cause the signal to appear. For example, if genes are only related to differentiation, then sub-clustering might not be necessary but could be of interest.
#See which genes cause variances in the data
get_var_genes(out_kidney, "UBCD")[,1:3]
#> Singular.vector.1 Singular.vector.2 Singular.vector.3
#> Highest-1 RPS6 HES1 CLU
#> Highest-2 DHRS2 FOS CTSH
#> Highest-3 SPINK1 ID2 MGST3
#> Highest-4 S100A6 ID1 EPCAM
#> Highest-5 HPGD JUN CYB5A
#> Highest-6 VSIG2 DDIT4 GSTM3
#> Highest-7 KRT7 JUNB CD9
#> Highest-8 FXYD3 DUSP1 GSTP1
#> Highest-9 FBLN1 GADD45B CD24
#> Highest-10 S100P HSP90AB1 TUBA4A
#> Highest-11 S100A11 ADIRF DDX5
#> Highest-12 ADIRF RGS16 SKP1
#> Highest-13 SNCG IER2 AGR2
#> Highest-14 PVALB FABP5 S100A11
#> Highest-15 UPK1A ID3 MYL6
#> Highest-16 UQCRQ TXNIP HSP90AB1
#> Highest-17 RPS18 H3F3A ENO1
#> Highest-18 HMGCS2 UBB TSPAN1
#> Highest-19 FTH1 HSPA1A ITM2B
#> Highest-20 PSCA RBP1 MYL12B
#> Highest-21 ADH1C GPC3 ARG2
#> Highest-22 RPL34 IGF2 CALM2
#> Highest-23 LGALS3 SPARC KRT8
#> Highest-24 RPL31 HSP90AA1 MYL12A
#> Highest-25 SHROOM1 TPM2 H3F3B
#> Highest-26 LEAP2 SNCG SYPL1
#> Highest-27 UPK2 TSC22D1 LGALS3BP
#> Highest-28 CISD3 HSPA1B KRT19
#> Highest-29 RPLP1 LGALS1 GAPDH
#> Highest-30 MT-ND3 SMC2 CLIC1
#> Highest-31 RPL12 HNRNPA2B1 RGS2
#> Highest-32 IGF2 MYLIP MALAT1
#> Highest-33 S100A4 CALD1 CAPG
#> Highest-34 PERP HMGB2 LINC00675
#> Highest-35 MT-ND4 NR2F1 AOC1
#> Highest-36 FABP5 YME1L1 GATA2
#> Highest-37 FBP1 COL1A2 SCPEP1
#> Highest-38 GDF15 NR2F2 TMEM176B
#> Highest-39 RPL26 SEPT7 ACTG1
#> Highest-40 MT-ATP6 IDH1 HSPA5
#> Highest-41 C9orf16 TMSB15A DEGS2
#> Highest-42 RPS14 ZNF503 UBC
#> Highest-43 RPL41 DNAJA1 CLDN7
#> Highest-44 FAM3B DDIT3 CAPS
#> Lowest-1 TMSB4X PHLDA2 RPS2
#> Lowest-2 NGFRAP1 AQP2 COL1A2
#> Lowest-3 ACTB TNFRSF12A COL3A1
#> Lowest-4 IGFBP7 KRT18 COL1A1
#> Lowest-5 WFDC2 ERRFI1 RPL13A
#> Lowest-6 CLDN3 RPL41 RPL13
#> Lowest-7 NDUFA4 HES4 PTN
#> Lowest-8 MEST SAT1 RPL28
#> Lowest-9 HINT1 CLU RPS3
You can also check out the fit of the MP distribution for each cluster.
#Check if the MP distribution fits to the data
plot_MP(out_kidney, "UBCD")

And for fruther validation, see if the singular vectors of the significant singular values look meaningful. By plotting either clusters or genes with the singular vectors.
#Plot clusters
plot_singular_vectors(out_kidney, "UBCD", colour = sce_kidney@metadata$ubcd.cluster)

#Plot variance driving genes
plot_singular_vectors(out_kidney, "UBCD", colour = "UPK1A", scaled = FALSE)

总而言之一句话,SMGMA值越接近于1,越包含有意义的subcluster,越要进行cluster的再分群分析。
生活很好,等你超越