scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
12-使用ArchR进行主题和功能丰富
鉴定出一个稳固的峰集后,我们通常希望预测哪些转录因子可能介导形成这些可接近的染色质位点的结合事件。这有助于评估标记峰或差异峰,以了解这些峰组是否富集了特定转录因子的结合位点。例如,我们经常在细胞类型特异性可及的染色质区域中发现关键的谱系定义TF。以类似的方式,我们可能要测试各种峰组以丰富其他已知特征。例如,我们可能想知道细胞类型A的特定于细胞类型的ATAC-seq峰是否丰富了另一组基因组区域,例如ChIP-seq峰。本章详细介绍了如何在ArchR中执行这些扩充。
12.1微分峰中的母峰富集
继续上一章对差异峰的分析,我们可以寻找在各种细胞类型中向上或向下的峰中富集的基序。为此,我们必须首先将这些主题注释添加到我们的中ArchRProject。这有效地创建了一个二进制矩阵,其中每个峰中都有一个基序以数字表示。我们使用addMotifAnnotations()确定存储在中的峰集中的基序是否存在的功能来执行此操作ArchRProject。
projHeme5 <- addMotifAnnotations(ArchRProj = projHeme5, motifSet = "cisbp", name = "Motif")
然后,我们可以使用上一章中生成的差异测试SummarizedExperiment对象markerTest来定义一组我们对基序富集测试感兴趣的差异峰。在这种情况下,我们正在寻找具有FDR <= 0.1和的峰Log2FC >= 0.5。在进行差异比较的情况下markerTest,这些代表峰在“类红细胞”细胞中比“祖细胞”细胞更易接近。我们可以使用该peakAnnoEnrichment()功能测试这些差异可访问的峰,以丰富各种图案。该功能是可推广的功能,可用于许多不同的浓缩测试,我们将在本章中进行演示。
motifsUp <- peakAnnoEnrichment(
seMarker = markerTest,
ArchRProj = projHeme5,
peakAnnotation = "Motif",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
的输出peakAnnoEnrichment()是一个SummarizedExperiment包含多个对象的对象,这些对象assays存储了利用超几何检验进行的浓缩测试的结果。
motifsUp
为了准备用于绘制的数据,ggplot我们可以创建一个简化的data.frame对象,其中包含图案名称,校正后的p值和重要性等级。
df <- data.frame(TF = rownames(motifsUp), mlog10Padj = assay(motifsUp)[,1])
df <- df[order(df$mlog10Padj, decreasing = TRUE),]
df$rank <- seq_len(nrow(df))
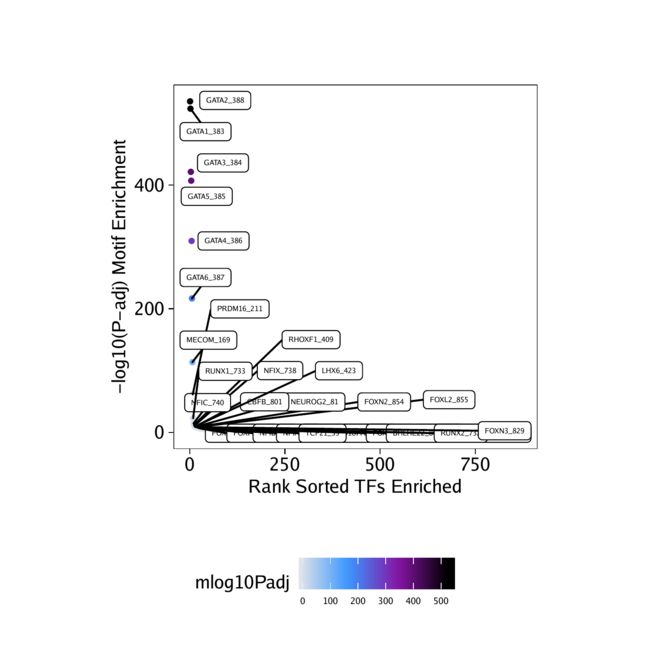
如预期的那样,在“类红细胞”细胞中更易于接近的峰中最富集的基序与GATA转录因子相对应,与GATA1在类红细胞分化中的深入研究一致。
head(df)
使用ggplot我们可以绘制按顺序排列的TF主题,并根据其丰富的重要性对其进行着色。在这里,我们ggrepel用来标记每个TF主题。
ggUp <- ggplot(df, aes(rank, mlog10Padj, color = mlog10Padj)) +
geom_point(size = 1) +
ggrepel::geom_label_repel(
data = df[rev(seq_len(30)), ], aes(x = rank, y = mlog10Padj, label = TF),
size = 1.5,
nudge_x = 2,
color = "black"
) + theme_ArchR() +
ylab("-log10(P-adj) Motif Enrichment") +
xlab("Rank Sorted TFs Enriched") +
scale_color_gradientn(colors = paletteContinuous(set = "comet"))
ggUp
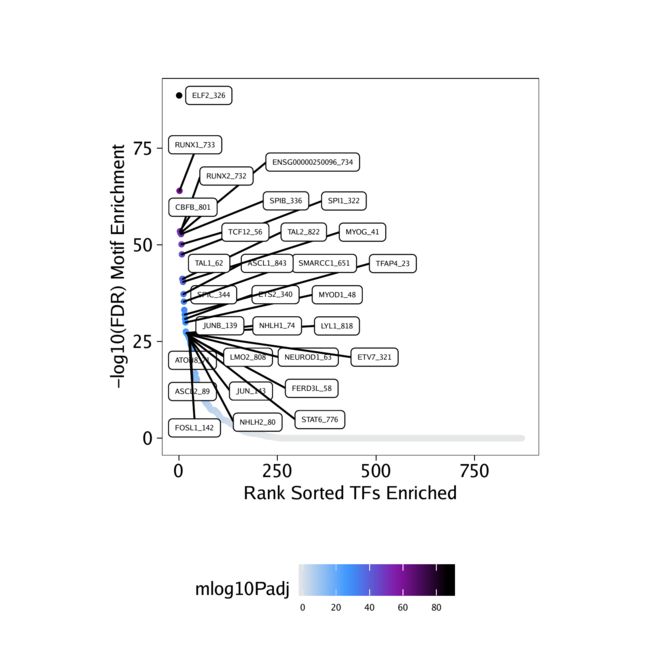
通过使用带有的峰,我们可以对“祖细胞”单元中更易于访问的峰执行相同的分析
Log2FC <= -0.5。
motifsDo <- peakAnnoEnrichment(
seMarker = markerTest,
ArchRProj = projHeme5,
peakAnnotation = "Motif",
cutOff = "FDR <= 0.1 & Log2FC <= -0.5"
)
motifsDo
df <- data.frame(TF = rownames(motifsDo), mlog10Padj = assay(motifsDo)[,1])
df <- df[order(df$mlog10Padj, decreasing = TRUE),]
df$rank <- seq_len(nrow(df))
在这种情况下,“祖细胞”中更易接近的峰中最富集的基序对应于RUNX,ELF和CBFB基序。
head(df)
ggDo <- ggplot(df, aes(rank, mlog10Padj, color = mlog10Padj)) +
geom_point(size = 1) +
ggrepel::geom_label_repel(
data = df[rev(seq_len(30)), ], aes(x = rank, y = mlog10Padj, label = TF),
size = 1.5,
nudge_x = 2,
color = "black"
) + theme_ArchR() +
ylab("-log10(FDR) Motif Enrichment") +
xlab("Rank Sorted TFs Enriched") +
scale_color_gradientn(colors = paletteContinuous(set = "comet"))
ggDo
要保存这些图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(ggUp, ggDo, name = "Erythroid-vs-Progenitor-Markers-Motifs-Enriched", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE)
12.2标记峰的母体富集
与上一部分中对差分峰进行的基序富集分析相似,我们还可以对使用识别的标记峰进行基序富集getMarkerFeatures()。
为此,我们将标记的峰值SummarizedExperiment(markersPeaks)传递给peakAnnotationEnrichment()函数。
enrichMotifs <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "Motif",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
的输出peakAnnoEnrichment()是一个SummarizedExperiment包含多个对象的对象,这些对象assays存储了利用超几何检验进行的浓缩测试的结果。
enrichMotifs
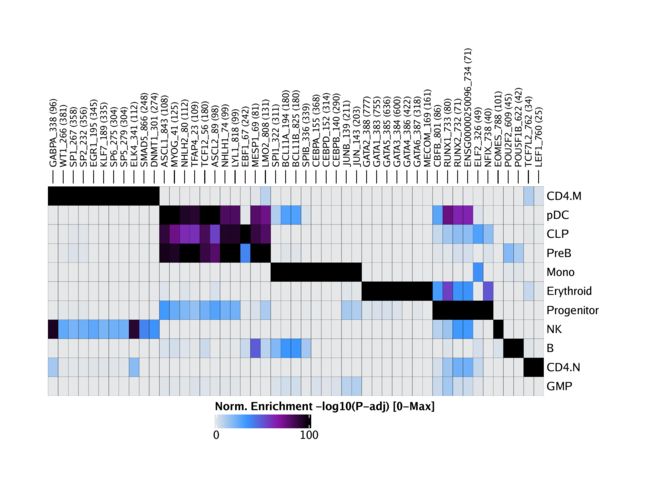
我们可以使用该plotEnrichHeatmap()函数在所有细胞组中直接绘制这些基序的富集。在此功能中,我们使用n参数限制每个单元格组显示的图案总数。
heatmapEM <- plotEnrichHeatmap(enrichMotifs, n = 7, transpose = TRUE)
我们可以使用来显示该情节ComplexHeatmap::draw()。
ComplexHeatmap::draw(heatmapEM, heatmap_legend_side = "bot", annotation_legend_side = "bot")
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(heatmapEM, name = "Motifs-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
12.3 ArchR富集
除了测试用于富集图案的峰外,ArchR还可以确定更多可定制的富集。为了促进这一级别的数据探索,我们精心挑选了一些不同的功能集,可以轻松对其进行测试,以丰富您感兴趣的峰值区域。我们将在下面描述每个精选功能集。这种分析最初是受LOLA启发的。
12.3.1编码TF绑定站点
ENCODE协会已将TF结合位点(TFBS)映射到了广泛的细胞类型和因子中。我们可以使用这些TFBS集合更好地了解我们的集群。例如,在真正未知的细胞类型的背景下,这些富集可以帮助阐明细胞身份。要使用这些ENCODE TFBS功能集进行分析,我们只需使用调用该addArchRAnnotations()函数collection = "EncodeTFBS"。与使用时类似addPeakAnnotations(),这将创建所有标记峰与所有ENCODE TFBS之间的重叠的二进制表示。
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "EncodeTFBS")
然后,我们可以使用peakAnnoEnrichment()函数通过峰集测试这些ENCODE TFBS的富集。
enrichEncode <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "EncodeTFBS",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
如前所述,此函数返回一个SummarizedExperiment对象。
enrichEncode
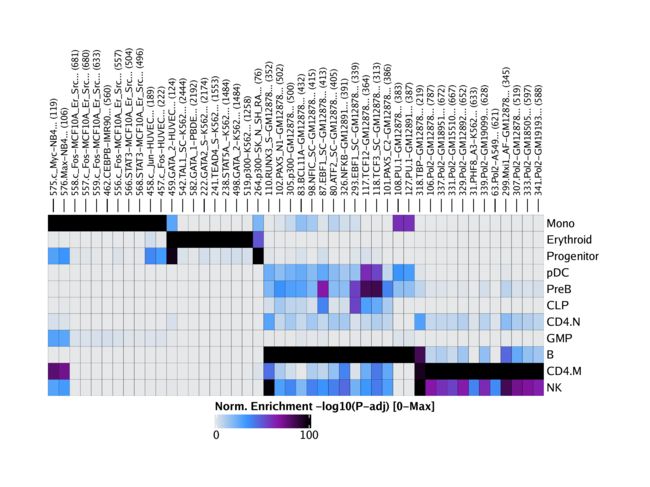
我们可以使用plotEnrichHeatmap()函数根据这些富集结果创建一个热图。
heatmapEncode <- plotEnrichHeatmap(enrichEncode, n = 7, transpose = TRUE)
然后使用绘制该热图ComplexHeatmap::draw()。
ComplexHeatmap::draw(heatmapEncode, heatmap_legend_side = "bot", annotation_legend_side = "bot")
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(heatmapEncode, name = "EncodeTFBS-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
12.3.2批量ATAC序列
与精选的ENCODE TF结合位点类似,我们还精选了来自批量ATAC-seq实验的峰调用,可用于重叠富集测试。我们可以通过设置访问这些批量ATAC-seq峰集collection = "ATAC"。
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "ATAC")
然后,我们通过设置来测试标记峰以富集这些大量的ATAC-seq峰peakAnnotation = "ATAC"。
enrichATAC <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "ATAC",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
和以前一样,此输出是SummarizedExperiment具有有关浓缩结果信息的对象。
enrichATAC
我们可以SummarizedExperiment使用来创建一个浓缩热图plotEnrichHeatmap()。
heatmapATAC <- plotEnrichHeatmap(enrichATAC, n = 7, transpose = TRUE)
并使用绘制此热图 ComplexHeatmap::draw()
ComplexHeatmap::draw(heatmapATAC, heatmap_legend_side = "bot", annotation_legend_side = "bot")
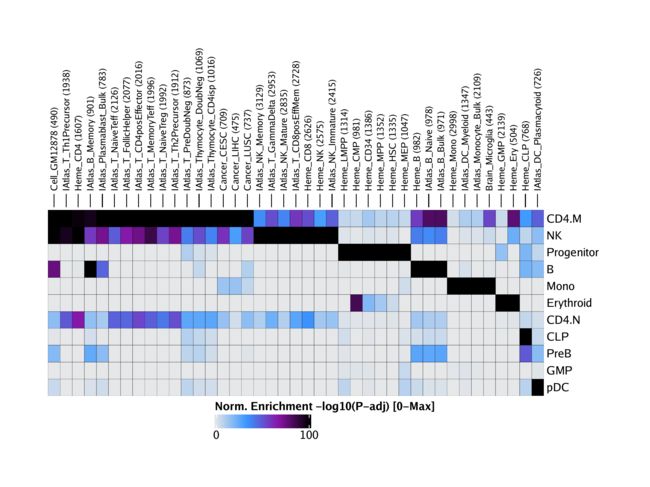
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(heatmapATAC, name = "ATAC-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
12.3.3法典TFBS
通过设置可以对CODEX TFBS 执行相同类型的分析collection = "Codex"。
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "Codex")
enrichCodex <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "Codex",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
enrichCodex
heatmapCodex <- plotEnrichHeatmap(enrichCodex, n = 7, transpose = TRUE)
ComplexHeatmap::draw(heatmapCodex, heatmap_legend_side = "bot", annotation_legend_side = "bot")
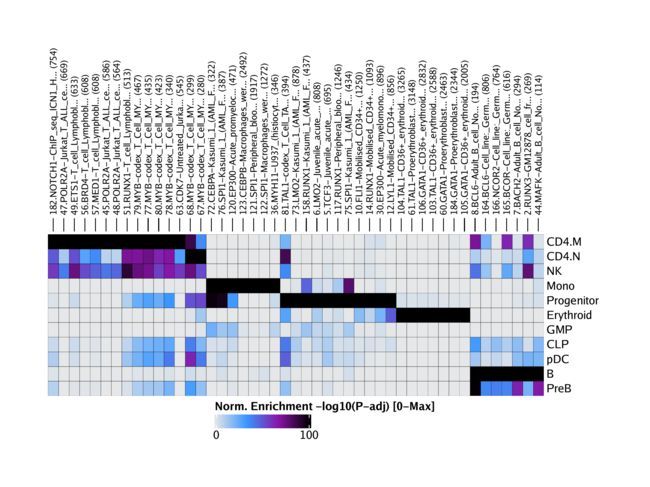
然后我们可以绘制这个
plotPDF(heatmapCodex, name = "Codex-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
12.4定制丰富
除了所有这些精选注释集之外,ArchR还能够接受用户定义的注释以执行自定义扩充。在以下示例中,我们说明了如何基于选择的ENCODE ChIP-seq实验创建自定义注释。
首先,我们将定义将要使用的数据集并提供下载链接。本地文件可以以相同的方式使用。
EncodePeaks <- c(
Encode_K562_GATA1 = "https://www.encodeproject.org/files/ENCFF632NQI/@@download/ENCFF632NQI.bed.gz",
Encode_GM12878_CEBPB = "https://www.encodeproject.org/files/ENCFF761MGJ/@@download/ENCFF761MGJ.bed.gz",
Encode_K562_Ebf1 = "https://www.encodeproject.org/files/ENCFF868VSY/@@download/ENCFF868VSY.bed.gz",
Encode_K562_Pax5 = "https://www.encodeproject.org/files/ENCFF339KUO/@@download/ENCFF339KUO.bed.gz"
)
然后,我们ArchRProject使用addPeakAnnotation()函数向我们添加自定义注释。在这里,我们将自定义注释称为“ ChIP”。
projHeme5 <- addPeakAnnotations(ArchRProj = projHeme5, regions = EncodePeaks, name = "ChIP")
和以前一样,我们使用此自定义批注使用来执行峰批注富集,peakAnnoEnrichment()并按照相同的步骤创建批注热图。
enrichRegions <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "ChIP",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
enrichRegions
heatmapRegions <- plotEnrichHeatmap(enrichRegions, n = 7, transpose = TRUE)
ComplexHeatmap::draw(heatmapRegions, heatmap_legend_side = "bot", annotation_legend_side = "bot")
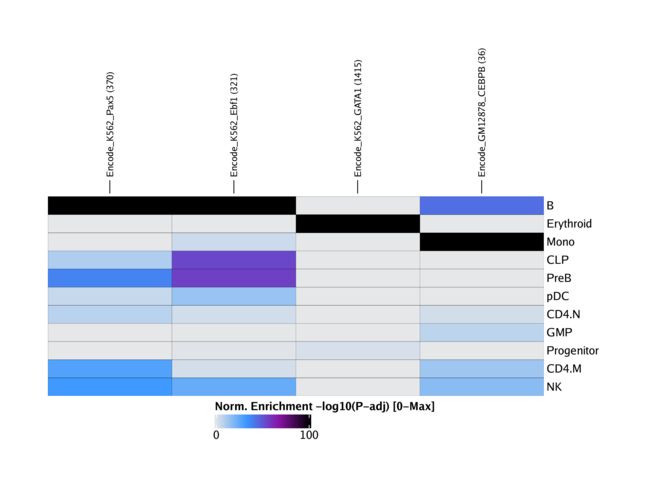
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(heatmapRegions, name = "Regions-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
参考材料:
https://www.archrproject.com/