基于Gromacs的蛋白分子动力学模拟(RMSD、RMSF及蛋白的回旋半径)
一、实验要求
实验对象:目标体系为modeller或其他方法建模的结果中评价最好的模型。
软件: Gromacs-5.1.2
二、实验步骤
- 加立场
gmx pdb2gmx –h 打开帮助菜单。 选力场的时候选择 Amber99sb…,溶剂类型选Tip3p。
2、加模拟盒子,溶剂层厚度为0.8nm。
gmx editconf -bt ( boxtype: 做三个盒子的对比cubic/triclinic/dodecahedron ), -d 0.8 , 比较三种类型的盒子水分子数目差别。
3、加水溶剂。
gmx solvate –h
4、做能量优化。 参数文件:em.mdp(可从官网上下载) etol: 500,达到收敛,该步骤完成。
5、平衡体系,将体系升温。 从0K升温到300K,在30ps内完成。
6、动力学模拟采样。模拟时间:1ns,步长:2fs。坐标保存的频率为每10ps保存一帧结果,整个轨迹共100个frame.
7、结果分析:
7.1. 全体系的alpha-C原子的均方根偏差(RMSD)结果获取及分析,gmx rms 。
7.2. 全体系的alpha-C原子的均方根涨落(RMSF)结果获取及分析, gmx rmsf。
7.3. 体系的总势能变化曲线分析,采用g_energy命令。
7.4. 分析蛋白质的回旋半径变化,采用g_gyrate命令
7.5. 将采样最后的构象与初始构象进行叠加比较,分析构象的变化情况。
7.7. 从模拟的轨迹中将体系中的蛋白质单独取出来,另存为一个轨迹文件protein.xtc, 用VMD的插件”movie maker”做成一个小电影。
三、操作过程记录及结果
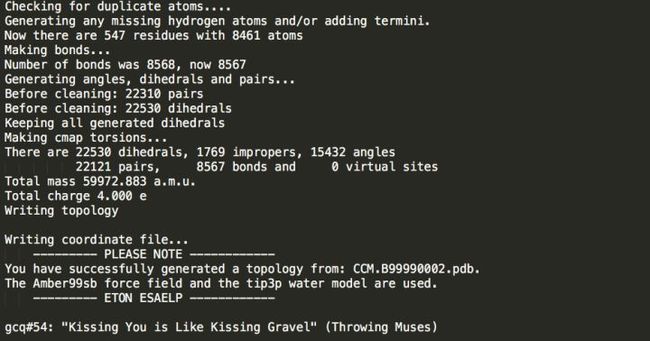
Step1: 输入如下命令,在命令行的交互式操作中,选择力场(输入5)AMBER99SB protein,选择溶剂模型(输入1):TIP3P
gmx pdb2gmx -f CCM.B99990002.pdb -o conf.gro -p topol.top
选择力场和溶剂模型后出现如下屏幕回显(部分):
Step2:加盒子。输入如下命令:分别加三个模拟盒子,比较三种类型的盒子水分子数目差别
gmx editconf -f conf.gro -bt cubic -d 1.0 -o cubic_out.gro
gmx editconf -f conf.gro -bt triclinic -d 1.0 -o triclinic_out.gro
gmx editconf -f conf.gro -bt dodecahedron -d 1.0 -o dodecahedron_out.gro
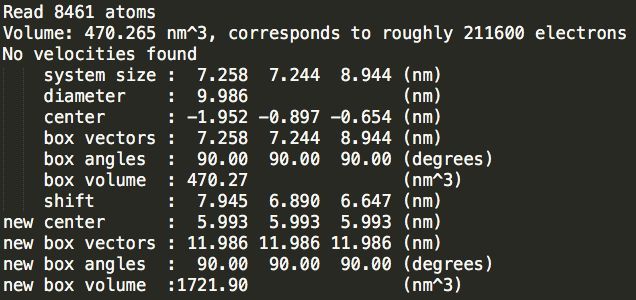
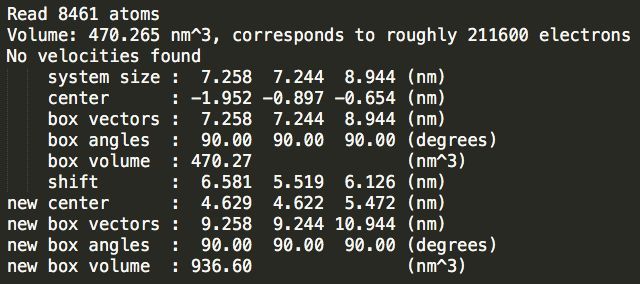
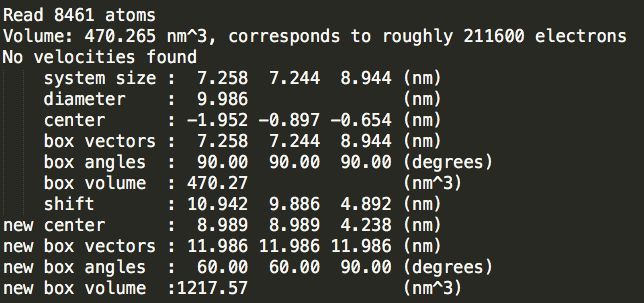
Step3:加溶剂。
gmx solvate -cp cubic_out.gro -o cubic.pdb -p topol.top
gmx solvate -cp triclinic_out.gro -o triclinic.pdb -p topol.top
gmx solvate -cp dodecahedron_out.gro -o dodecahedron.pdb -p topol.top


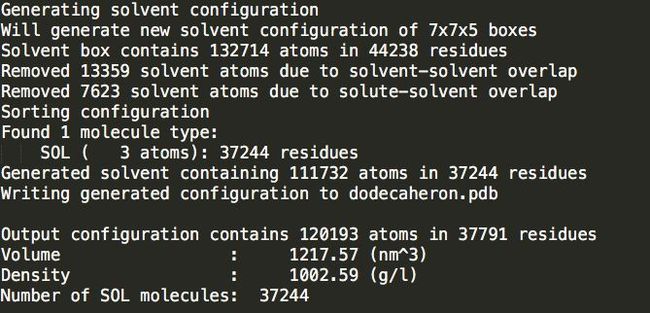
cubic_out.gro
triclinic_out.gro
dodecahedron_out.gro
53895
27862
37244
可以发现立方体盒子里水分最多。
Step4:做能量优化, etol: 500,达到收敛,该步骤完成。
从官网上下载一个em.mdp的例子文件,下载地址如下
http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin/gmx-tutorials/complex/Files/em.mdp 检测离子平衡
输入如下命令,检测体系的离子平衡:
gmx grompp -f em.mdp -c cubic.pdb -p topol.top -o cubic_em
检测结果如下:发现需要添加四个负离子

Step5:添加离子,发现总共是4个正电荷,需要加4负电荷 添加离子命令:
gmx genion -s cubic_em.tpr -o ION.gro -p topol.top -nn 4 -pname NA 选择(SOL)
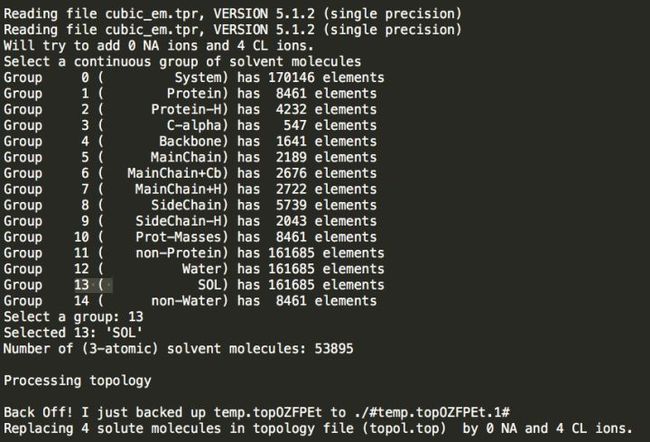
加离子后:
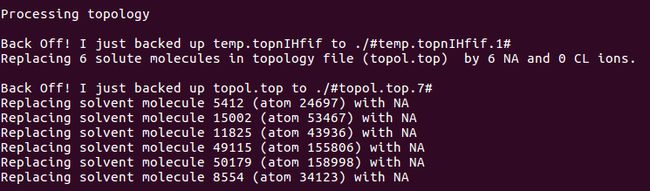
Step6:再次检测离子平衡,使用新的文件ION.gro进行检测:
gmx grompp -f em.mdp -c ION.gro -p topol.top -o cubic_em.tpr
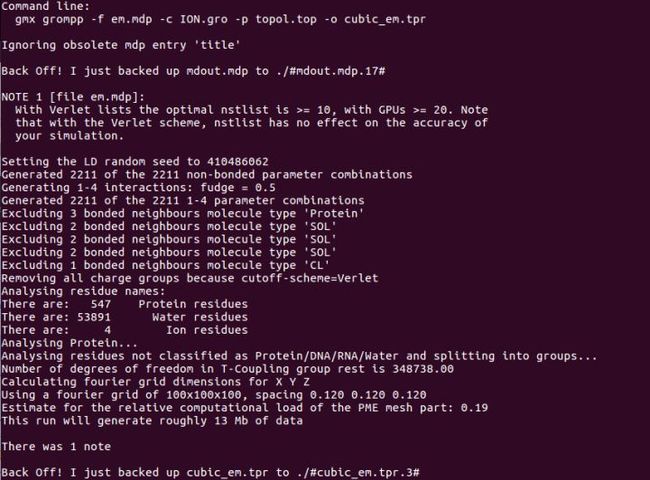
能量最小化:
gmx mdrun -s cubic_em.tpr -deffnm cold -v

能量查看(potential)
gmx energy -f cold.edr -o cold.xvg

将cold.xvg导入Excel绘制能量曲线图,看能量是否降低,如下图:
可以发现能量确实降低了。

Step7:平衡体系,将体系升温。 从0K升温到300K,在30ps内完成
根据生成的mdout.mdp文件,创建uprade.mdp,将体系升温至300k,步长为15000,改步长:
![]()
gmx grompp -f upgrade.mdp -c cold.gro -p topol.top -o cubic_em_hot.tpr 回显如下:

gmx mdrun -s cubic_em_hot.tpr -v -deffnm hot 回显如下:

然后进行能量查看(选择tempratrue):
gmx energy -f hot.edr -o hot.xvg 回显如下:

将hot.xvg导入excel表格发现确实升到了300k

Step6: 体系采样
gmx grompp -f sample.mdp -c hot.gro -p topol.top -o cubic_em_hot_sample.tpr

sample.mdp需要根据老师要求进行修改,修改地方如下:
模拟时间:1ns,步长:2fs。坐标保存的频率为每10ps保存一帧结果,整个轨迹共100个frame
输入命令:gmx mdrun -s cubic_em_hot_sample.tpr -v -deffnm hot_sample

能量查看(temperature),能量确实达到了300K(约等于)
gmx energy -f hot_sample.edr -o hot_sample.xvg

Step7:结果分析与讨论
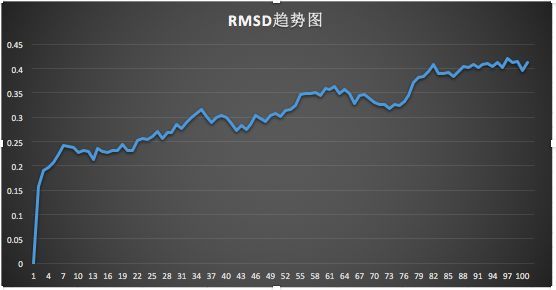
1、全体系的alpha-C原子的均方根偏差(RMSD)结果获取及分析
输入如下命令:gmx rms -s cubic_em_hot_sample.tpr -f hot_sample.trr -o rmsd.xvg

RMSD值可以反应出模拟过程中体系的稳定情况,将rmsd.xvg导入EXCEL表,绘折线图,由图可知动力学模拟逐渐达到平衡。
- 全体系的alpha-C原子的均方根涨落(RMSF)结果获取及分析
输入如下命令:gmx rmsf -s cubic_em_hot_sample.tpr -f hot.trr -o rmsf.xvg

RMSF计算每个原子相对于其平均位置的涨落, 表征了结构的变化对时间的平均, 给出了蛋白各个区域柔性的表征, 对应于晶体学中的b因子(温度因子). 通常, 我们预期RMSF和温度因子类似, 这可以用于考察模拟结果是否与晶体结构符合。

3、体系的总势能变化曲线分析
gmx energy -f hot_sample.edr -o potential.xvg

可以看出总势能总体在波动 :

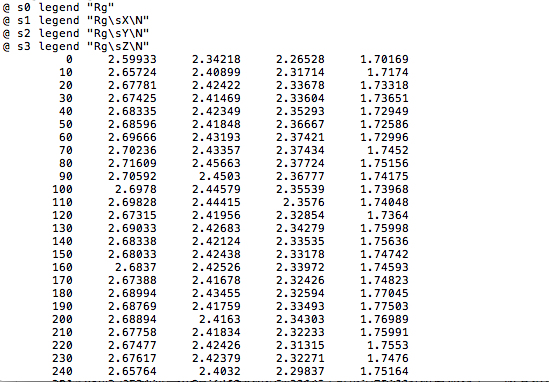
4、分析蛋白质的回旋半径变化(只绘制了半径的曲线图)
gmx gyrate -f hot_sample.trr -s cubic_em_hot_sample.tpr -o gyrate.xvg

回旋半径是描述蛋白质紧密型的一个物理量,半径越小说明致密性越好,,即蛋白质结构就越稳定。
分析蛋白质的回旋半径变化(只绘制了半径的曲线图),如图第一列是蛋白质的回旋半径:
由图可看出蛋白质的回旋半径有逐渐变小。

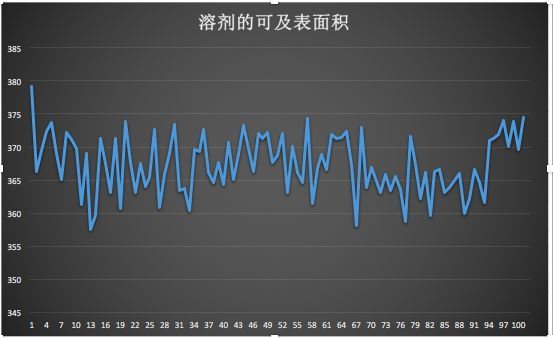
5、分析溶剂的可及表面积(SASA)选择SOL
gmx sasa -f hot_sample.trr -s cubic_em_hot_sample.tpr -o area.xvg
它是描述蛋白质疏水性的重要手段,氨基酸残基的疏水性是影响蛋白质折叠的重要物理作用

6、将采样最后的构象与初始构象进行叠加比较,分析构象的变化情况
gmx confrms -f1 ION.gro -f2 hot_sample.gro -o fit.pdb
感觉用肉眼看不太出来,变化不太大。

Step8:VMD小电影制作
用最新生成的liuwanlin.pdb做vmd小电影。
导入pdb——选择Extension->visualization->movie maker,Movie duration(seconds)设置生成视频的总时长,设定20s,使用录屏软件录小视频。
最后,有需要欢迎通过微信公众号联系我们。
微信公众号:320科技工作室。