用R语言做WGCNA分析全步骤一(附代码解读)【转载】
代码逐句分析
- 一、文章来源
- 二、基因共表达网络构建及模块识别
-
- 1.数据导入、清洗及预处理
- 2.检查过度缺失值和离群样本
- 3.聚类做离群样本检测
- 4.载入临床特征数据
- 三、自动构建网络及识别模块
-
- 1.确定合适的软阈值:网络拓扑分析
- 2.一步构建网络和识别模块
一、文章来源
初学WGCNA,觉得博主的代码写的很不错,但其中很多代码在第一遍看的时候有很多地方不理解,后查阅了很多资料,终于看明白了,于是写了一篇笔记,记录自己的学习心得,有不准确的地方,还望各位大佬们不吝赐教~
原文链接:WGCNA简明指南
二、基因共表达网络构建及模块识别
1.数据导入、清洗及预处理
示例数据下载:https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/FemaleLiver-Data.zip
打开后是这样的,一共包括三个文件,依次是:
①临床性状文件,里面有样本名,各性状数据,其他无用数据
②基因注释文件,包含基因ID与基因名之间的对应关系,方便后面转换
③基因表达矩阵,样本名以及各基因的表达量构成的表达矩阵
三个文件可以自己打开看一下

注:如果library(‘WGCNA’)出现问题,请转链接:WGCNA包安装
# BiocManager::install("WGCNA")
library('WGCNA')
# 在读入数据时,遇到字符串之后,不将其转换为factors,仍然保留为字符串格式
options(stringsAsFactors = FALSE)
# 导入示例数据,这里填写自己存放表达矩阵的路径
femData = read.csv("D:\\desktop\\FemaleLiver-Data\\LiverFemale3600.csv") # femData代表该文件
# 查看数据
dim(femData) # dim查看矩阵形状
names(femData) # names查看列标,即每一列的标题
*** 此时femData存放的是带有其他信息的各样本的基因表达量
如下图,行标MMT…表示基因名,列标F2_2这种表示样本名,但还有其他的无用的列,所以需要把这些列删除
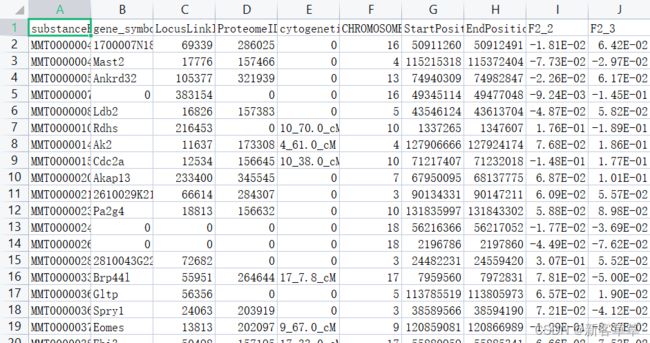
# 提取样本-基因表达矩阵
datExpr0 = as.data.frame(t(femData[, -c(1:8)]))
# 删除femData矩阵第1到8列,再转置,再变为数据格式,将所得用datExpr0表示
# 第一列是基因名,也删除了,即要以所有列标都是样本名的形式做转置
names(datExpr0) = femData$substanceBXH
# 转置后,行标变成了样本名,然后重新加入基因名作为列标
# femData$substanceBXH表示在femData矩阵里面依次取substanceBXH列的值
# names表示列标,即将这些值(基因名)作为datExpr0的列标
rownames(datExpr0) = names(femData)[-c(1:8)]
# 将femData列标的第1到8个删除后,其余的列标依次作为datExpr0的行标,但本步可以不用,因为在定义datExpr0时就已经完成了这不
***names是列标,rownames是行标
至此,我们得到的datExpr0是一个列表为样本名,行标为基因的一个基因表达矩阵,按如下代码观察其前六行六列
> datExpr0[1:6,1:6]
MMT00000044 MMT00000046 MMT00000051 MMT00000076 MMT00000080 MMT00000102
F2_2 -0.0181000 -0.0773 -0.02260000 -0.00924 -0.04870000 0.17600000
F2_3 0.0642000 -0.0297 0.06170000 -0.14500 0.05820000 -0.18900000
F2_14 0.0000644 0.1120 -0.12900000 0.02870 -0.04830000 -0.06500000
F2_15 -0.0580000 -0.0589 0.08710000 -0.04390 -0.03710000 -0.00846000
F2_19 0.0483000 0.0443 -0.11500000 0.00425 0.02510000 -0.00574000
F2_20 -0.1519741 -0.0938 -0.06502607 -0.23610 0.08504274 -0.01807182
2.检查过度缺失值和离群样本
不解释,直接按下面格式,输入对应矩阵名字即可
# 检查缺失值太多的基因和样本
gsg = goodSamplesGenes(datExpr0, verbose = 3);
gsg$allOK
如果最后一个语句返回TRUE,则所有的基因都通过了检查。如果没有,我们就从数据中剔除不符合要求的基因和样本。
不返回TRUE就直接运行下面代码
if(!gsg$allOK)
{
#(可选)打印被删除的基因和样本名称:
if(sum(!gsg$goodGenes)>0)
printFlush(paste("Removinggenes:",paste(names(datExpr0)[!gsg$goodGenes], collapse =",")));
if(sum(!gsg$goodSamples)>0)
printFlush(paste("Removingsamples:",paste(rownames(datExpr0)[!gsg$goodSamples], collapse =",")));
#删除不满足要求的基因和样本:
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
3.聚类做离群样本检测
我们是对离群样本做检测,目标是样本,所以行标得是样本名,故用我们上面处理得到矩阵datExpr0
目的是通过样本的聚类树看看是否有任何明显的异常值。
sampleTree = hclust(dist(datExpr0), method ="average");
# dist()表示转为数值,method表示距离的计算方式,其他种类的详见百度
sizeGrWindow(12,9)
# 绘制样本树:打开一个尺寸为12 * 9英寸的图形输出窗口
# 可对窗口大小进行调整
# 如要保存可运行下面语句
# pdf(file="Plots/sampleClustering.pdf",width=12,height=9);
par(cex = 0.6) # 控制图片中文字和点的大小
par(mar =c(0,4,2,0)) # 设置图形的边界,下,左,上,右的页边距
plot(sampleTree, main ="Sample clustering to detectoutliers",sub="", xlab="", cex.lab = 1.5,
cex.axis= 1.5, cex.main = 2)
# 参数依次表示:sampleTree聚类树,图名,副标题颜色,坐标轴标签颜色,坐标轴刻度文字颜色,标题颜色
# 其实只要包括sampleTree和图名即可
画出的树如图所示
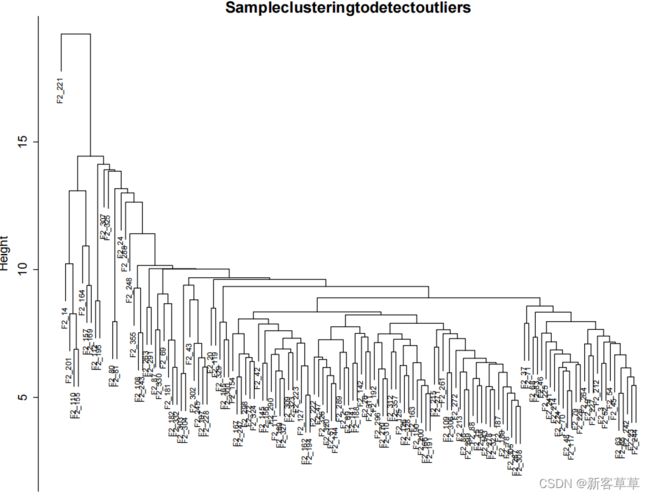 可见有一个异常值。可以手动或使用自动方法删除它。选择一个**高度(height)**进行切割将删除异常样本,比如15(Fig1b),并在该高度使用分支切割
可见有一个异常值。可以手动或使用自动方法删除它。选择一个**高度(height)**进行切割将删除异常样本,比如15(Fig1b),并在该高度使用分支切割
# 绘制阈值切割线
abline(h = 15,col="red"); # 高度15,颜色红色
# 确定阈值线下的集群
clust = cutreeStatic(sampleTree, cutHeight = 15, minSize = 10)
# 以高度15切割,要求留下的最少为10个
table(clust) # 查看切割后形成的集合
# clust1包含想要留下的样本.
keepSamples = (clust==1) # 将clust序号为1的放入keepSamples
datExpr = datExpr0[keepSamples, ]
# 将树中内容放入矩阵datExpr中,因为树中剩余矩阵不能直接作为矩阵处理
nGenes =ncol(datExpr) # ncol,crow分别表示提取矩阵的列数和行数
nSamples =nrow(datExpr)
输入table(clust),得到的图片是

***datExpr是去除离群样本后,剩下的样本及其基因表达
下面是其前六行六列

4.载入临床特征数据
将样本信息与临床特征进行匹配。
traitData =read.csv("D:\\desktop\\FemaleLiver-Data\\ClinicalTraits.csv")
dim(traitData) # 看看形状
names(traitData) # 看看列标
# 删除不必要的列.
allTraits = traitData[, -c(31, 16)] # 将去掉第31和16列(两个不包含数据的列)后的traitData存入allTraits中
allTraits = allTraits[,c(2, 11:36) ] # 在allTraits中保留第2,11到36列(只取样本名的性状相关数据)
# 这时的allTraits是只包含样本名和性状相关数据的矩阵
dim(allTraits) # 看看形状
names(allTraits) # 看看列标
# 形成一个包含临床特征的数据框
femaleSamples =rownames(datExpr) # femaleSamples存放存放录入基因表达量的样本名称
traitRows =match(femaleSamples, allTraits$Mice) # 将表达矩阵和性状矩阵中,样本名(Mice)重复的这些样本在allTraits中的行标返回给traitRows(一个数字向量)
datTraits = allTraits[traitRows, -1] # 在allTraits中取上步的得到的这些行(行),并删除第一列,然后组成矩阵datTraits
rownames(datTraits) = allTraits[traitRows, 1] # 因为上一步删了第一列,所以重新赋予第一列,即这些行的样本名字
collectGarbage() # 释放内存
***traitData是各样本的临床性状
***allTraits是只包含样本名和性状相关数据的矩阵
***datTraits表示样本中每个性状的表达情况
将临床特征与样本树状图的关系可视化。
sampleTree2 = hclust(dist(datExpr), method ="average")
# 重新聚类样本
traitColors = numbers2colors(datTraits, signed = FALSE);
# 将临床特征值转换为连续颜色:白色表示低,红色表示高,灰色表示缺失
plotDendroAndColors(sampleTree2, traitColors,
groupLabels =names(datTraits),
main ="Sample dendrogram and trait heatmap")
# 在样本聚类图的基础上,增加临床特征值热图
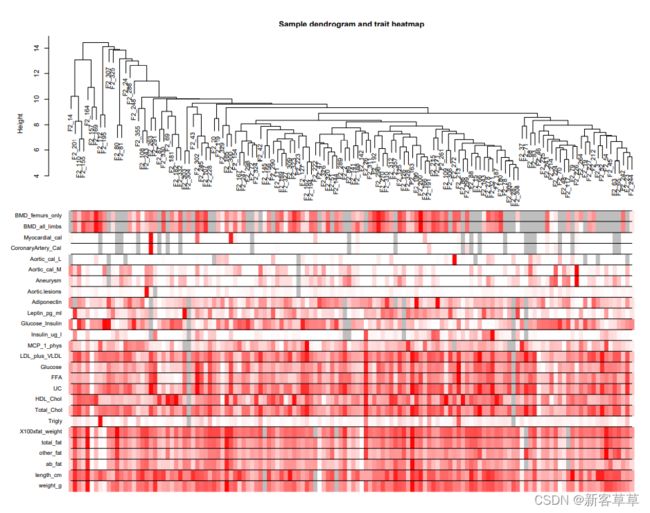
三、自动构建网络及识别模块
1.确定合适的软阈值:网络拓扑分析
软阈值的目的:为了衡量两个基因是否具有相似表达模式,一般需要设置阈值来筛选,高于阈值的则认为是相似的。WGCNA分析时采用相关系数加权值,即对基因相关系数取N次幂,使得网络中的基因之间的连接服从无尺度网络分布(在某一复杂系统中,大部分节点只有少数几个连结,而某些节点却拥有与其他节点的大量连接),更具有生物意义。
下面这步会用就行,比较流程化,不求甚解
# 设置软阈值调参范围,powers是数组,包括1,2,...10,12,14,...,20
powers =c(c(1:10),seq(from = 12, to=20,by=2))
# 网络拓扑分析
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
# 绘图
sizeGrWindow(9, 5) # 图片的宽度和高度
# 1行2列排列
par(mfrow =c(1,2)); # 一页多图,一页被分为一行,两列
cex1 = 0.9;
# 无标度拓扑拟合指数与软阈值的函数(左图),下面的会用就行
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="SoftThreshold(power)",ylab="ScaleFreeTopologyModelFit,signedR^2",type="n",
main =paste("Scaleindependence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
# 这条线对应于h的R^2截止点
abline(h=0.90,col="red")
# Mean Connectivity与软阈值的函数(右图)
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="SoftThreshold(power)",ylab="MeanConnectivity", type="n",
main =paste("Meanconnectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5],labels=powers, cex=cex1,col="red")
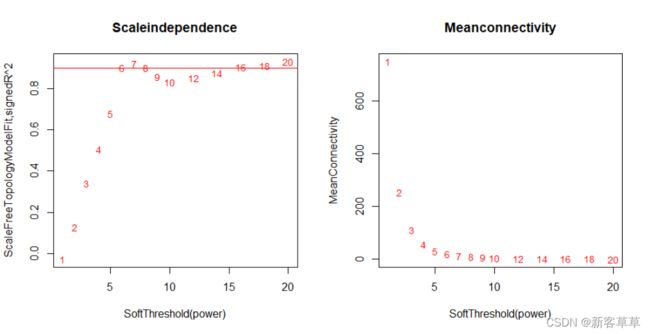 找第一个大于横线的power值,发现是6,那么软阈值就是6
找第一个大于横线的power值,发现是6,那么软阈值就是6
2.一步构建网络和识别模块
把所有的基因分为不同的基因模块
下面代码中未注释的参数就不管了
net = blockwiseModules(datExpr,power= 6, # 表达矩阵,软阈值
TOMType ="unsigned", minModuleSize = 30, # 数据为无符号类型,最小模块大小为30
reassignThreshold = 0, mergeCutHeight = 0.25, #mergeCutHeight合并模块的阈值,越大模块越少
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase ="femaleMouseTOM",
verbose = 3)
deepSplit 参数调整划分模块的敏感度,值越大,越敏感,得到的模块就越多,默认是2;
minModuleSize 参数设置最小模块的基因数,值越小,小的模块就会被保留下来;
mergeCutHeight 设置合并相似性模块的距离,值越小,就越不容易被合并,保留下来的模块就越多
这时的net里就已经包含了基因分类为模块的相关信息了
 表明有18个模块,按大小递减顺序标记为1到18,大小从609到34个基因。标签0为所有模块之外的基因。
表明有18个模块,按大小递减顺序标记为1到18,大小从609到34个基因。标签0为所有模块之外的基因。
# 可视化模块
sizeGrWindow(12, 9)
# 将标签转换为颜色
mergedColors = labels2colors(net$colors)
# 绘制树状图和模块颜色图
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Modulecolors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
下图除灰色外,每种颜色都代表着一种基因模块,即里面的基因功能是相似的
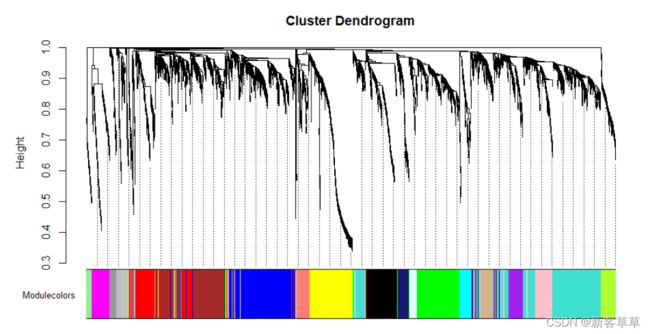
保存模块赋值和模块特征基因信息,以供后续分析。
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs;
geneTree = net$dendrograms[[1]];
save(MEs, moduleLabels, moduleColors, geneTree,
file="FemaleLiver-02-networkConstruction-auto.RData")
如下图,moduleLabels代表每个基因对应的模块序号(在基因名下面)
 如下图,moduleLabels代表每个基因对应的模块所属颜色(略去了基因名,但顺序同上图一样)
如下图,moduleLabels代表每个基因对应的模块所属颜色(略去了基因名,但顺序同上图一样)
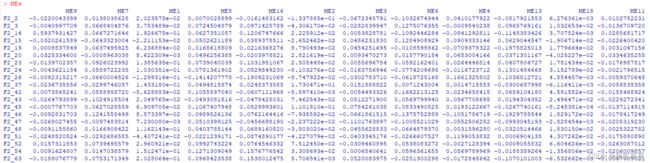
 如下图,MEs是以基因模块为单位,各个样本在这个模块中的表达量(别管是怎么来的)
如下图,MEs是以基因模块为单位,各个样本在这个模块中的表达量(别管是怎么来的)