【Scanpy】单细胞转录组分析思路之细胞分类和组织分层
本文将详细讲述单细胞转录组分析的步骤。
一、文章信息
题目:ClusterMap for multi-scale clustering analysis of spatial gene expression
链接:https://doi.org/10.1038/s41467-021-26044-x
期刊:Nature
二、数据集
数据集:小鼠初级视觉皮层V1_1020
转录组技术:STARmap
基因数:1020
细胞数:1650
总表达数:471295
细胞类型:16种
三、单细胞测序工作
关于这一部分,这篇文章没有讲解,我在了解生物背景的时候,查阅了一些资料,关于单细胞最初的制备工作。

对于这篇文章,在作者给的公开代码和数据中,数据已经是处理好的有关所有基因点的三维位置坐标。
四、细胞分类
进行细胞分类的关键是得到有细胞位置的数据集。
1、输入文件
文件由三部分组成,X,obs,var。三者的位置关系如下图所示:
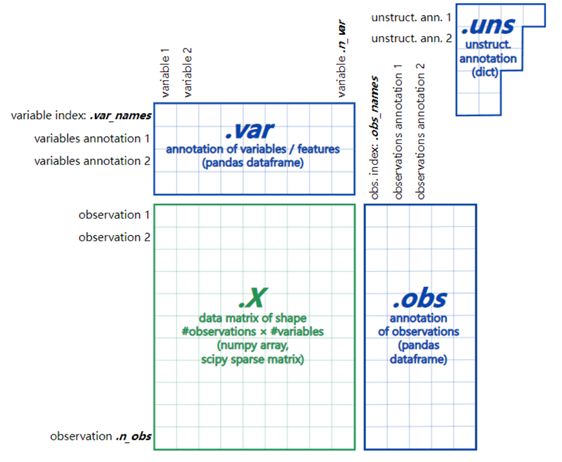
输入文件,得到adata数据格式,但是下文的操作都在细胞层面,所以对应查看adata.obs文件。
expr_path = os.path.join(out_path, 'expr_BY1.csv') # 里面全是0 1 2 3 的数字
var_path = os.path.join(out_path, 'var_BY1.csv') # 基因的名称
obs_path = os.path.join(out_path, 'obs_BY1.csv') # 二维行列坐标
# add expression data to the AnnData object 添加表达式数据到AnnData对象
expr_x = np.loadtxt(expr_path, delimiter=',') # delimiter表示分隔符,加载文件的分隔符
var = pd.read_csv(var_path, header=None) #header=None(文件中不包含列名的行)
var = pd.DataFrame(index=var.iloc[:,0].to_list())
obs = pd.read_csv(obs_path, index_col=0)
adata = AnnData(X=expr_x, var=var, obs=obs)2、预处理
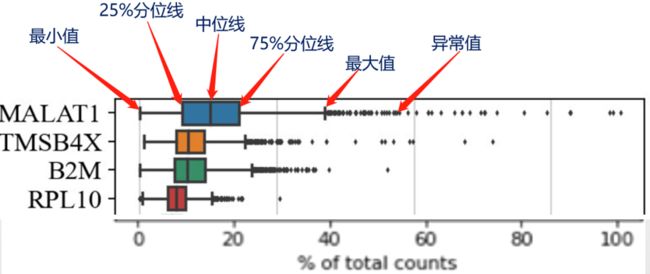
(1)可视化高表达基因
sc.pl.highest_expr_genes(adata, n_top=20) 
可视化所有细胞中表达量最多的20个基因,展示的方法采用的是箱线图。箱线图是对每个基因在所有细胞中表达量分布的更详细描述。箱子的宽度,反应了数据的波动程度,箱子越窄,数据分布越集中。
箱线图的查看以下图为例:中位线表示的是MALAT1在单个细胞表达量的中位数;中位数对应的是该基因表达量占该细胞总基因表达量的17%。可见,MALAT1基因在大部分细胞中的表达量占比约在10%~20%之间。
(2)计算质量控制指标
sc.pp.calculate_qc_metrics(adata, percent_top=None, inplace=True)在这里inplace=True,会将得到的细胞、基因质量控制指标添加到adata.obs和adata.var中。详细的添加的质量指标,如下文所示。其中:


(3)可视化基因表达情况
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'],
jitter=0.4, multi_panel=True)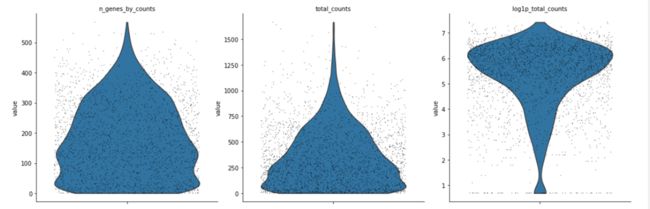
(4)直方图
su.plot_stats_per_cell(adata, save=False)查看表达情况:①表达数为这么多(横轴)的细胞有多少个(纵轴);②基因为这么多(横轴)的细胞有多少个(纵轴)。
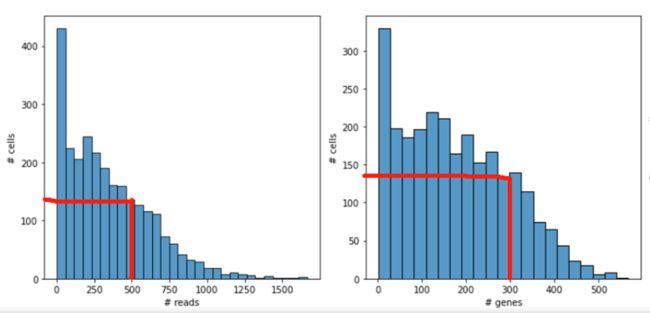
(5)散点图
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')每个点代表一个细胞。横轴:一个细胞中基因的总表达量;纵轴:一个细胞中表达的基因种类数。

(6)分位数
su.show_reads_quantile(adata)查看细胞中基因的总表达量。

(7)细胞过滤
根据直方图和分位数的结果,设定过滤指标。
sc.pp.filter_cells(adata, min_genes=5) # 过滤一个细胞中表达少于五个基因的细胞样本
sc.pp.filter_genes(adata, min_cells=5) # 过滤在少于五个细胞中表达的基因
sc.pp.filter_cells(adata, min_counts=29) # 过滤每个细胞中计数少于29个的细胞样本 至此,数据预处理部分结束,筛选合适的基因与数据,目的是为了给之后的分析提供更高质量的数据。
3、归一化处理
归一化的目的实际上就是把数据做一个转换,使同一基因在不同样本之间有可比性;同时降低数据的离散程度。在数据转换后都会变成log(x+1),使用log转换的一个好处,就是使得数据更加集中。
(1)归一化
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)对细胞进行归一化,一个细胞里的基因求和为1,这样一个基因的表达量就显而易见了,容易找到高表达量基因。
(2)存储数据
adata.raw = adata将 AnnData 对象的 .raw 属性设置为归一化和对数化的原始基因表达,以便以后用于基因表达的差异测试和可视化。这只是冻结了 AnnData 对象的状态。
(3)可视化高变基因
高变基因:在某些细胞里表达量高,但在其他较多细胞表达量较低。表达量差别较大。
特征选择的时候会用到高变基因。特征选择:对数据集基因进行过滤只保留对数据有贡献的基因。
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
sc.pl.highly_variable_genes(adata)
# 绘制特异性基因散点图
adata = adata[:, adata.var.highly_variable]
# 高变基因的选取
4、降维(PCA)
通过运行主成分分析 (PCA) 来降低数据的维数,力求用最少的维度去捕捉更多的数据特征。
sc.tl.pca(adata, svd_solver='arpack') # 通过主成分分析降低数据维度
sc.pl.pca_variance_ratio(adata, log=False)
# 检查单个 PC 对数据总方差的贡献,这可以提供给我们应该考虑多少个 PC 以计算细胞的邻域关系的信息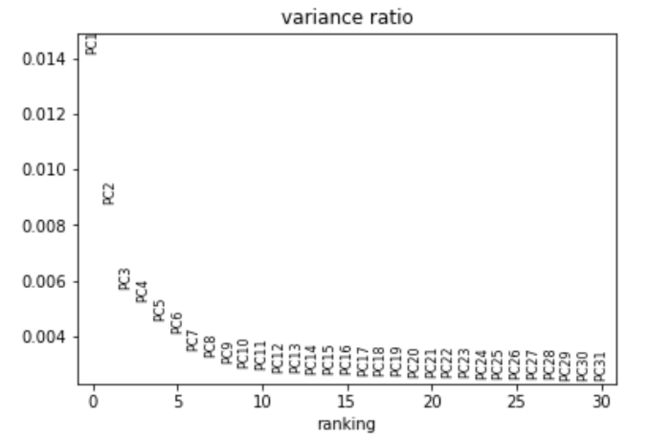
sc.pl.pca(adata, color='batch') #可视化PCA降维
5、聚类
首先使用数据的 PCA 表示来计算细胞的邻域图,之后使用UMAP可视化。
# Computing the neighborhood graph
sc.pp.neighbors(adata, n_neighbors=50, n_pcs=15)
# Run UMAP
sc.tl.umap(adata)
# Plot UMAP
sc.pl.umap(adata, color='batch')接下来使用louvain聚类,得到细胞类群。
# Run louvain cluster
sc.tl.louvain(adata, resolution=0.9) # resolution=0.9 提供的是分辨率 一般默认分辨率是1.0
# Plot UMAP with cluster labels
sc.pl.umap(adata, color='louvain') 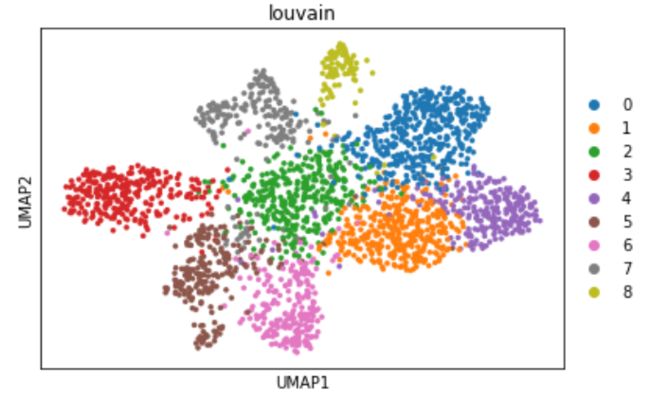
6、(分为三类)
在生物数据处理的过程中,很多时候是根据已知的生物特性,进行调整,在该文章中,对细胞按照神经元进行划分,可分为三类:兴奋神经元、抑制神经元、非神经元。所以接下来对已经分为的九类再重新划分为三类。在这里根据高表达基因进行划分。
top_markers = ["Gad1","Slc17a7"]
plt.figure(figsize=(30,15))
su.plot_heatmap_with_labels(adata, top_markers, 'louvain', show_axis=True, font_size=10)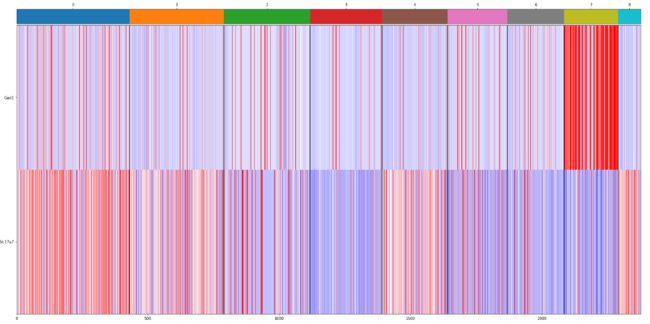
根据热图展示的表达情况,手动分为三类:把0、1、4、8合并;2,3,5,6合并。

未完待续……