RNA-seq Review:RNA-seq数据分析
文献:RNA-seq数据分析最佳实践调查
本次阅读 Genome Biology杂志 2016年Online的RNA-seq数据分析方法的 Review论文,题目为:
A survey of best practices for RNA-seq data analysis
本文翻译来自该文章。
RNA是基因组和蛋白组的中间体,因此转录本的鉴定和定量是重要的生物学问题。该论文综述了RNA-seq项目中相关的各个步骤、每个步骤的局限、和其他组学的整合以及展望。
Note : 从摘要中可以发现本文综述分为两部分(1)现有RNA-seq数据分析各个步骤的详细描述;(2)RNA-seq与多组学技术整合和未来展望。
摘要:
RNA-seq应用广泛,但是没有一种分析流程适用于所有情况。作者综述了RNA-seq数据分析的主要步骤,包括:实验设计、质量控制、读取比对、基因和转录水平的定量、可视化、差异基因表达、可变剪接、功能分析、基因融合检测和 eQTL 定位。作者强调了每个步骤的挑战,讨论了小 RNA 的分析以及 RNA-seq 与其他功能基因组学技术的整合。
最后,作者讨论了正在改变转录组学现状的新技术的前景。
(一)实验设计:(如Fig. 1)
要以回答生物学问题为导向设计RNA-seq实验,其中主要包含文库类型、测序深度和重复次数
1.提取RNA
(1)OligodT富集mRNA
对于真核生物,当样本量足够时,RNA中90%以上为rRNA,通常选择OligodT富集mRNA(1-2%尾部存在PolyA结构互补)。mRNA通常容易降解,需要衡量mRNA的RNA完整指数RIN (RNA integrity number)。
(2)去除rRNA
有些活检样本不足,富集的方法提取RNA效果差。此时,选择降解rRNA来得到mRNA。对于细菌等样本,因为是原核生物,mRNA不存在PolyA的结构,上述方法同样失效,也需要通过降解rRNA得到mRNA。
2.链特异型文库
通常Illumina测序的RNA-seq数据并未区分正链和负链,这使得反义转录和重叠表达的转录本定量无法区分。通常,链特异型文库通过dUTP标记方法,随后消化包含dUTP的链,以此得到特定链的RNA文库。
对于单端测序SE(single-end)和双端测序PE(paired-end),双端测序的文库更长,这样有利于转录本组装和转录本亚型(isoform)的鉴定。对于高质量的参考基因组,SE测序数据已经够用,PE测序数据对于注释不佳的基因组效果更好。发帖时(2021年),目前市场测序价格下降明显,PE数据效果更好,SE测序的应用越来越少了,已经逐渐淘汰。
3.测序深度或文库大小
最佳的测序深度取决于实验目的,通常测序深度越深定量结果越准,更多的鉴定到更多的转录本。有学者认为比对500万的reads已经足够定量中等表达和高表达的真核转录本,对于低表达的转录本需要1亿的reads。研究低复杂度的单细胞时,处理100万的reads,5万的reads已经足够定量高表达基因,甚至2万个reads可以区分组织细胞类型。
最佳的测序文库取决于目标实验的复杂度,实验证明增加测序深度确实可以改善转录本的鉴定和定量情况,但是可能会检测噪音或者脱靶转录本,饱和曲线可用于评估在给定测序深度下预期转录组覆盖率的改善。
4.样本重复次数
一个关键的设计因素是重复次数。要考虑技术性重复、生物学重复和统计效力(power),这些是功效分析的一部分。充分规划测序实验以避免技术偏差与良好的实验设计同样重要,尤其是当实验涉及大量样本需要分批处理时。在这种情况下,包括对照、随机样本处理和测序运行的智能管理对于获得正确的数据至关重要。详细数据可以参考Table1信息。


(二)RNA-seq数据分析
实际上RNA-seq的分析非常多样,此部分作者主要描述了典型RNA-seq数据分析的步骤,包括质量控制、有参和无参的数据比对、计算基因和转录本的表达量和差异表达基因鉴定。此外,作者也讨论了可变剪切、转录本融合和小RNA表达,最后讨论了可视化的工具包。
1.质量控制检查
RNA数据的获取包括几个部分:获取原始reads、reads比对和表达定量。
(1)原始reads质控
原始reads的质量控制涉及序列质量、GC含量、接头adapter、过表达的kmer和重复的reads(duplicated reads),通过这些来检测测序错误和PCR副产物和污染。可接受的重复,kmer和GC含量是物种特异性的,这些数据对于相同实验的样本应该保持同质。作者建议丢弃差异超过30%的异常值。
FastQC是应用于Illumina reads的,而NGSQC可以应用于任何平台。通常3’端的末端碱基的测序质量下降,如果太低,需要剔除这些碱基以提高比对效果。FASTX-Toolkit和Trimmomatic等软件工具可用于丢弃低质量reads、修剪接头序列并剔除低质量碱基。
(2)Read 比对情况衡量标准
Reads通常比对到基因组或转录组。
一个重要的参数是比对百分比,反映了整体的测序准确性和DNA污染情况。例如,期望的人类基因组中转录组的比对百分比应该为70%-90%(取决于比对软件),其中很大一部分reads均可以较好地比对到有限数量的相同区域(多比对reads,multi-mapping reads);当比对到人类转录组时,整体比对率会降低,因为未注释为转录本的序列将会丢失。此外,由于比对到转录本亚型(isoform)的共享的reads,导致多比对reads会增加。
另一个重要的参数是reads在外显子子和比对链上的覆盖均匀度。如果reads主要集中在PolyA选择的3’端,这可能说明RNA质量较低。比对中的reads可能存在GC含量可能反应PCR偏差,比对质量控制工具包括 Picard、RSeQC和 Qualimap`。
(3)定量
一旦计算了转录本实际的表达量值,就应该矫正GC含量和基因长度,以便矫正的归一化方法能应用。
若参考转录本注释质量高,可以预测样本的生物型组成,进一步反应RNA纯化步骤结果如何。比如rRNA和小RNA。许多 R 包(例如 NOISeq 或 EDASeq)为转录本的count table质量控制提供用图。
(4)样本间可重复性
上述质量控制步骤是基于个体的,检测重复之间的可重复性和批次效应来评估RNA-seq数据的整体质量也很重要。
技术性重复中斯皮尔曼相关性(Spearman, R2 > 0.9)要大于0.9,没有明确的生物学重复标准,因为实验系统存在异质性。若不同的实验条件下存在差异表达基因,那么PCA结果中的相同实验条件下的样本期望聚类在一起。
(三)转录本鉴定
当存在参考基因组和参考转录本时,RNA-seq数据可以比对到这两个数据上来推断转录本的表达量。比对到注释好的基因组或转录本上,不能发现新的、未鉴定的转录本。
当不存在参考基因组等的参考文件时,首先策略是将转录组数据组装成长的contig片段,然后将这些contig作为表达的转录本,通过reads比对回来进行定量。
在以上两种情况下,read的覆盖度均能量化转录本的表达水平,区别是转录本的鉴定和定量是顺序进行还是同时进行。
(1)唯一比对和多重比对
无论比对到基因组还是转录本,reads都可能是唯一比对(uniquely mapped)或者多重比对(multi-mapped)。多重比对是因为重复序列或者基因组内的旁系同源基因中保守domain的存在。当比对基因组时,多重比对占据重要部分,这些reads不应该被丢弃。当比对转录本时,转录本亚型(isoform)中共享的外显子会增加多重比对。 对于存在可变剪切的基因而言,两种比对策略均面临转录本鉴定和定量的挑战。
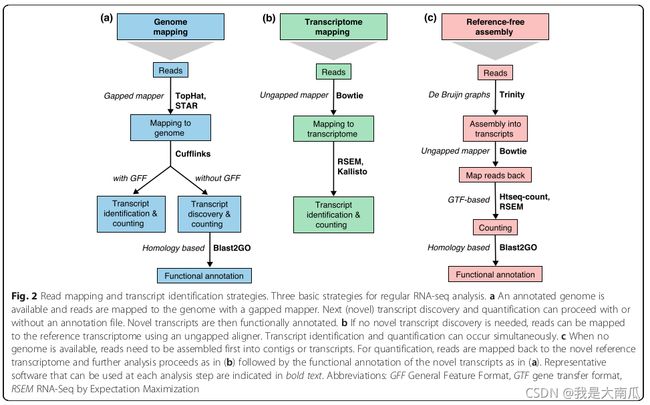
(2)转录本鉴定
Illumina测序reads短,跨几个剪切位点的概率小,因此推断转录本全长困难。
注释转录起始和终止位点困难,借助数据库有更可能正确的注释转录本亚型(isoform)(软件GRIT,数据库CAGE和RAMPAGE)。
双端reads(PE)和更深的测序覆盖度有助检测低表达转录本,重复有助于检测假阳性信号。Cufflinks、iReckon、SLIDE 和 StringTie,通过将现有注释添加到可能的同种亚型(isoform)列表中来合并现有注释。Augustus软件可以用来注释蛋白编码基因,对非编码转录本效果较差。 总之,短reads重构转录本难度较大,多种方法之间分歧也很大。
(3)转录本重头预测
当物种没有公布的数据可用时,需要重头组装转录本序列。使用的软件有SOAPdenovo-Trans, Oases,Trans-ABySS或者Trinity。 通常双端reads(PE)或长reads效果更好,因为他们包含更多的信息。缺乏足够覆盖的低表达转录本很难预测,但是reads也不是越多越好,过多的reads会导致错误组装和运行时间增加。因此,对于深度测序的样本,需要计算机减少reads数。对于跨样本的比较分析,建议将样本合并,组装成较好的整合的转录本,再进行表达定量。
总之,无论有参考还是无参考的数据,短reads重构转录本都是相对困难的,结果会产生片段化的转录本,长度长表现效果更好,如Pacific Biosciences 的 SMRT。
(四)转录本定量方法
RNA-seq数据的常见应用是估计基因或者转录本的表达丰度。应用的最初是统计比对到转录本的reads数,即为count data,也有一些算法无需比对,如基于kmer计数的Sailfish软件。最简单的方法就是通过HTSeq-count或 featureCounts等软件得到比对数据的原始count值。基因水平的定量通常使用GTF文件,文件内包含外显子和基因的坐标,通常丢弃多重比对的reads。
(1)Reads原始Count的矫正方法
原始的read count在样本间不能比较,会受到转录本长度,reads数量和测序偏差的影响。RPKM值对特征长度(基因长度或转录本长度)及文库效应进行标准化。FPKM是RPKM的衍生值,对于SE数据,二者结果一致。针对PE reads,与CPM也相似。RPKM和FPKM都是样本内(within-sample)的标准化方法。TPM可以通过FPKM转换而来,现在更常用。在文献中,样本内和样本间基因表达的比较存在一定的混乱。
对于一个基因在多个样本中的表达量比较,无需进行基因长度的矫正。若对一个样本内不同的基因进行表达量的鉴定和排序,那么需要矫正基因长度,因为基因长度越长在捕获reads的时候更有优势。Cufflinks软件可以展现出不同基因长度对表达量的影响,目前,通常认为基因的TPM值更适合多样本间的平行比较,其中也存在问题,可以使用TMM归一化方法解决。
(2)表达量计算的软件
目前,已经存在几种复杂的算法来解决转录本水平共享reads的不同转录本之间的表达量。Cufflinks软件使用期望最大法来估算比对结果中转录本的表达量(e.g. TopHat 结果)。Cufflinks方法考虑了reads在基因长度上的非均匀分布,可以通过PE reads进行已有转录本和重头预测转录本的定量。
从转录组比对结果定量的算法包括 RSEM(通过期望最大化的 RNA-Seq)、eXpress、Sailfish和 kallisto等。这些方法在转录本和输出样本内归一化值之间分配多重比对reads,并针对测序偏差进行了校正。此外,RSEM 算法使用返回 TPM 值的期望最大化方法。 NURD软件是使用 SE reads估计转录表达的有效方法,具有低内存和低计算成本的特点。
(五)差异表达基因分析
差异表达分析要求基因的表达量在样本间具有可比性。
RPKM、FPKM 和 TPM 将比较样本的最重要因素标准化掉了,即测序深度,无论是直接还是通过说明转录本的数量,样本之间可能存在显著差异。这些方法依赖于基于总计数或有效计数的归一化方法,并且在样本间转录本分布不同时往往表现不佳,也就是说,当高度和差异表达的特征会改变样本内count的分布情况。考虑到这一点的归一化方法是 TMM、DESeq、PoissonSeq 和 UpperQuartile软件,它们忽略了高度可变或高度表达的特征(基因、转录本)。
干扰样本内比较的其他因素包括样本或条件之间转录本长度的变化、转录本覆盖范围的位置偏差(在Cufflinks中说明)、平均片段大小以及基因(在 EDAseq 包中更正)。NOISeq R 包包含各种各样的诊断图,用于识别 RNA-seq 数据中的偏差来源,并进行调整。此外,若存在批次效应,应该使用COMBAT或 ARSyN软件来消除。
RNA-seq的表达量是基于绝对计数的read count或概率分配的read count。因此计算差异表达的第一种方法使用离散概率分布,例如泊松或负二项式 [48, 54]。负二项式分布(也称为 gamma-Poisson 分布)是 广义化的Poisson 分布,允许 RNA-seq 数据特征的分子池中随机取样的预期方差之外的额外方差(称为过度离散,overdispersion)。
然而,只要考虑到小read count的抽样方差,就不需要使用离散分布来准确分析差异表达(对于少量重复的实验最重要)。 与上述离散分布方法相比,在了解数据方差组成的同时转换 RNA-seq 读数归一化计数的方法已被证明表现良好。 此外,经过大量的归一化(包括 TMM 和批次删除)后,数据可能会失去其离散性,更类似于连续分布。
一些方法,例如流行的 edgeR,将原始读取计数作为输入,并将可能的偏差源引入统计模型,以执行集成归一化和差异表达分析。在其他方法中,差异表达需要事先对数据进行归一化以消除所有可能的偏差。DESeq2 与 edgeR 一样,使用负二项式作为参考分布,并提供自己的归一化方法。 baySeq和 EBSeq是贝叶斯方法,也基于负二项式模型,它们定义了一组模型来描述实验组之间的差异并计算每个基因的每个实验组的后验概率。其他方法包括数据转换方法,该方法考虑了小读取计数的采样方差,并创建了可以通过常规线性模型进行分析的离散基因表达分布。
最后,非参数方法如 NOISeq或 SAMseq对数据做出最小的假设,并仅从实际数据中估计用于推理分析的零分布。对于比较没有或很少重复的两个样本的小规模研究,负二项式分布的估计可能是杂乱的。在这种情况下,基于泊松分布(例如 DEGseq)或经验分布(NOISeq)的更简单的方法可以是一种替代方法。应该强调的是,在没有生物复制的情况下,无法进行总体推断,因此任何 p 值计算都是无效的。因此,无需重复分析 RNA-seq 数据的方法仅具有探索性价值。考虑到测序价格的下降,当样本可用性不受限制时,我们建议 RNA-seq 实验至少进行三个生物学重复,以允许所有差异表达方法利用重复之间的可重复性。
最近的独立比较研究表明,方法的选择(甚至软件包的版本)可以显着影响分析结果,并且没有一种方法可能对所有数据集都表现良好。因此,我们建议彻底记录所用程序的设置和版本号,并考虑使用多个包进行重要分析的重复。
(六)可变剪切分析
转录本水平的差异表达分析可以检测转录本亚型的表达变化,并且已经提出了RNA-seq可变剪切的算法。主要分为两大类:
第一类:将亚型表达估计和差异表达检测整合到一起来估计一个基因中每个亚型的表达比例变化。早期方法,BASIS,可以推断差异表达的转录本亚型,而CuffDiff2首先估计亚型表达,然后比较差异。通过整合这两个步骤,在进行统计分析以寻找差异亚型表达时考虑了第一步中的不确定性 。流程差异度量 (flow difference metric,FDM) 使用来自比对的外显子读数和连接读数的对齐累积转录图来推断亚型,并使用 Jensen-Shannon 散度来测量差异。rSeqDiff是新提出的方法。
这些方法都受read短的内在限制。
第二类:基于外显子的方法,跳过了亚型预测,通过比较exon和junction的reads分布来推断比较样本中的可变剪切信号。该方法的前提是可以在外显子及其连接点(junction)的信号中跟踪亚型(isoform)表达的差异。DEXseq和 DSGSeq采用类似的思想,通过测试基因外显子(和junction)读数计数的显着差异来检测差异剪接基因。rMATS 通过比较用连接read定义的外显子包含水平来检测外显子的差异使用情况。rDiff 通过比较基因替代区域的read count来检测差异亚型表达,无论是否有注释的可变亚型。 DiffSplice 使用比对图(alignment graphs)来识别可变剪切模块 (ASM),并使用 ASM 的信号识别差异拼接。外显子或连接方法的优势在于它们在识别单个可变剪接事件方面具有更高的准确性。如果研究的重点不是整个多种亚型(whole isoforms),而是包含和排除特定外显子和它们包含的功能蛋白质域(或调节特征,如果是非翻译区外显子),则基于外显子的方法是合适的。
(七)RNA-seq数据中的可视化
RNA-seq 数据的可视化一般来说,类似于任何其他类型的基因组测序数据,它可以在读取级别完成(例如使用 ReadXplorer)或在处理的覆盖范围(读取堆积)、未标准化(例如,总计数)或标准化的水平上,使用基因组浏览器,例如 UCSC 浏览器、Integrative Genomics Viewer (IGV)、基因组图谱或 Savant。一些可视化工具是专门为可视化多个 RNA-seq 样本而设计的,例如 RNAseqViewer,它提供了灵活的方式来显示外显子、转录本和连接点上的读取丰度。可以隐藏内含子以更好地显示外显子上的信号,热图可以帮助对多个样本的信号进行视觉比较。实际上,RNAseqViewer 比 IGV 慢。
一些用于差异基因表达分析的软件包(例如 Bioconductor 中的 DESeq2 或 DEXseq)具有使结果可视化的功能,而其他软件包则是为可视化专用目的而开发的,例如 CummeRbund(用于 CuffDiff )或Sashimi图,可用于可视化差异剪接的外显子。Sashimi图的优势在于,当样本数量较少时,它们的连接读数显示更直观且美观。可以使用 SplicePlot获得用于可变剪切数量性状基因座 (sQTL) 的Sashimi 、结构和hive图。可以使用 SpliceSeq生成可变剪切图,并且 SplicingViewer绘制拼接连接点和替代拼接事件。 TraV 是一种集成数据分析的可视化工具,但其分析方法不适用于大型基因组。
总之,转录组的复杂性,多层信息的有效显示仍然是一个挑战。目前的工具对于单个基因的研究很有价值,仍然可以检查数据、评估结果准确性和解释生物学问题。也可以检测一些问题,如 3’ 偏差或复杂的转录结构。使用者需要各取所需地去可视化,研究和探索。
(八)基因融合事件的发现
染色体重排产生的融合基因发现和可变剪切的发现类似,但增加了搜索挑战,因为不再假设转录片段在单个染色体上是共线的。
即使使用最先进的工具,结果叶未必尽如人意。产物主要是由于多态性、同源性和测序错误导致的读取序列未对齐造成的。同源基因家族和高度多态性基因(如 HLA 基因)产生的reads无法轻松地唯一映射到它们在参考基因组中的起源位置。对于具有非常高表达的基因,RNA-seq 中不可忽略的测序错误率会产生错误地映射到同源基因座的读数。建议过滤高度多态性的基因和同源基因对。还建议过滤不太可能参与基因融合的高表达基因,例如核糖体RNA 。最后,融合边界附近嵌合与野生型读数的低比率可能表明来自高表达基因。
成功预测嵌合体后,下一步优先需要考虑的是生物学功能的融合基因,他们会引起基因组变异。被reads通读的嵌合体需要控制数据过滤或者不相关数据的识别。强融合序列的预测结果中每个子序列可以与单个融合基因高度特异性比对在一起。PE序列比SE效果更好。预测指标的另一个指标是剪切位点,对于多数融合基因,基因组短点位于内含子中,融合边界与剪切位点重合。此外,融合亚型通常遵循野生型基因的剪接模式。因此,高置信度预测具有与外显子边界一致的融合边界以及与野生型外显子匹配的外显子。融合鉴定工具会考虑上面的这些特征。
(九)小RNA
二代测序技术可以解决小RNA生物学问题,sRNA 的长度通常为 18-34 个核苷酸,包括 miRNA、短干扰 RNA (siRNA)、PIWI 相互作用 RNA (piRNA) 和其他类别的调节分子。复杂性低,测序reads通常2-10 million reads。
sRNA-seq 数据的生物信息学分析不同于标准 RNA-seq 协议(图 1c)。首先修剪连接的接头序列并计算所得的读取长度分布。在动物中,通常有 22 和 23 个核苷酸的峰值,而在植物中,有 21 和 24 个核苷酸冗余读数的峰值。例如,miRTools 2.0是一种用于预测和分析 sRNA 物种的工具,默认使用 18-30 个碱基长的读数。阈值取决于应用,在 miRNA 的情况下,通常在 19-25 个核苷酸的范围内。与标准 RNA-seq 一样,sRNA 读数必须使用标准工具(例如 Bowtie2、STAR或 Burrows-Wheeler Aligner-BWA)与参考基因组或转录组序列进行比对。然而,有一些比对器(例如 PatMaN [99] 和 MicroRazerS)已被设计用于比对具有适合短读段最佳比对的设置的短序列。比对可以设置有无错配,无错配更常用。
对于超过预期数量的比对reads,可以移除来自重复序列的部分。miRNA在每个基因组中通常有5-20个比对,随后计算表达。通过sRNA可以验证mRNA污染,比如检测是否存在高表达基因(如 GAPDH 或 ACTB)的异常read 覆盖度问题。
sRNA分析分为已知注释和重头预测。例如分别用于动物和植物 miRNA 的 miRDeep和 miRDeep-P ,或者 UEA sRNA Workbench上的反式作用 siRNA 预测工具。如 miRTools 2.0、ShortStack和 iMir等工具也可用于对 sRNA 文库进行综合注释和识别不同类别的 sRNA。
(十)RNA-seq功能解析
差异基因的功能或分子途径的注释是RNA-seq分析的最后一步。一方面可以通过DEG基因寻找功能,另一方面可以通过GSEA来排序,得到差异基因。
例如GOseq估算基因长度的偏差效应,并应用超几何分布来进行功能富集解释这种偏差。类似地,基因集变异分析 (GSVA)或 SeqGSEA包也结合了剪接并实施类似于 GSEA 的富集分析。功能分析需要为所研究的转录组提供足够的功能注释数据。 Gene Ontology、Bioconductor、DAVID或 Babelomics等资源包含大多数模型物种的注释数据。然而,在从头转录组组装或重建过程中发现的新转录本至少会缺乏一些功能信息,因此注释对于这些结果的功能分析是必要的。通过在蛋白质数据库(例如 SwissProt)和包含保守蛋白质结构域(例如 Pfam和 InterPro)的数据库中搜索相似的序列,可以使用直向学对蛋白质编码转录物进行功能注释。
使用标准词汇表,如基因本体论 (GO),允许跨直向同源物进行功能信息的一些交换。 Blast2GO 等流行工具允许针对各种数据库和受控词汇表对完整的转录组数据集进行大量注释。重构的转录本中50-80%的部分也可以进行Term语义注释,但是有些缺乏蛋白编码能力,保守型略差,Rfam 数据库包含大多数特征明确的 RNA 家族,例如核糖体或转移 RNA,而 mirBase或 Miranda专门研究 miRNA。这些资源可用于短非编码 RNA 的基于相似性的注释,但尚无标准功能注释程序可用于其他类型的 RNA,例如长非编码 RNA。
(十一)与其他数据的整合
RNA-seq数据可以作为输出结果,和后面的生物学研究紧密结合。
RNA 和 DNA 测序数据的组合。例如SNP和基因的表达相关联,可以鉴定eQTL,大型 eQTL 研究表明,遗传变异影响大多数基因的表达。相比于微阵列,RNA-seq数据在eQTL分析上有两个优势:一方面可以检测鉴定影响转录本表达的变异;其次,重叠杂合 SNP 的读数可以映射到母本和父本染色体,从而能够量化个体内的等位基因特异性表达。等位基因的特异性信号提高了关联定位的能力,这对计算性能是很大的消耗,许多软件只看顺式区域的SNP来优化分析。RNA-seq数据可以结合冲测序的SNP数据,剔除融合基因的假阳性个体,鉴定拷贝数变异。
RNA和甲基化数据的结合。通常成对的看甲基化模式和差异表达基因。这样鉴定的统计显著相关是小部分基因,可以借助网络互作,鉴定协调甲基化差异和RNA-seq差异的模块。
RNA和转录因子的ChIP-seq数据的结合。可以剔除ChIP-seq分析中的假阳性,证明TF对靶基因的激活或抑制作用。
微小RNA
RNA-seq 和 miRNA-seq 数据的整合有可能揭示 miRNAs 对转录稳态水平的调节作用。然而,这种分析具有挑战性,因为 miRNA 目标预测的噪声非常大,这阻碍了基于 miRNA 与其目标基因之间相关性的分析。可以在 mirWalk 和 miRBase 等数据库中找到关联,这些数据库根据各种算法提供目标预测。 CORNA、MMIA、MAGIA和 SePIA等工具通过测试基因、miRNA、通路和 GO 术语之间的显着关联,或通过测试相关性或反相关性来改进预测靶基因和相关miRNA的表达谱。通常,我们建议使用由多种算法预测的 miRNA-mRNA 关联。例如,在小鼠中,我们发现在五个数据库中需要 miRNA-mRNA 关联导致每个 miRNA 有大约 50 个目标 mRNA 预测(STATegra 观察)。
蛋白质组学和代谢组学
RNA-seq 与蛋白质组学的整合是有争议的,因为这两个测量结果通常显示出低相关性(~0.40 [158, 159])。 尽管如此,蛋白质组学和 RNA-seq 的成对整合可用于识别新的同种型。 未报告的肽可以从 RNA-seq 数据中预测,然后用于补充通常在质谱中查询的数据库。 此外,如果 RNA-seq 数据集的表达基因中不存在质谱分析中存在的肽,则可以识别翻译后编辑事件。 转录组学与代谢组学数据的整合已被用于识别在基因表达和代谢物水平上受调控的通路,并且可以使用在通路上下文中可视化结果的工具。
多种数据类型的集成和可视化
目前,多种数据的集成和可视化需要特别注意,因为每种分析有自己的流程和标准,需要详细了解清楚,更好地应用这些数据。
(十二)展望
RNA-seq 已成为转录组分析的标准方法,但技术和工具仍在不断发展。
当前 RNA-seq 应用的两个主要亮点是从少量起始材料构建转录组,以及从更长的读取中更好地识别转录本。
(1)单细胞RNA-seq
单细胞 RNA-seq (scRNA-seq) 是 RNA-seq 最新和最活跃的领域之一,具有独特的机遇和挑战。 Smart-seq和 Smart-seq2等较新的协议使我们能够从非常少量的起始 mRNA 开始工作,通过适当的扩增,可以仅从单个细胞中获得。由此产生的单细胞库能够识别组织中新的、未表征的细胞类型。
它们还使测量分子生物学中的一个迷人现象成为可能,即特定群体内其他相同细胞中基因表达的随机性。在这种情况下,单细胞研究只有在将一组单个细胞文库与细胞群进行比较时才有意义,目的是识别具有不同表达基因组合的多个细胞的亚群。少量的起始材料和 PCR 扩增限制了单细胞文库可以高效测序的深度,通常低于一百万个reads。由于细胞中单个 mRNA 分子的数量很少,更深入的测序可能发现和测量特异性表达的等位基因,因为额外的reads可以提供有用的证据。
单细胞转录组通常包括大约 3000-8000 个表达的基因,这远少于相应汇集种群的转录组中的数量。面临的挑战是将由于在单分子水平缺乏敏感性而导致的技术噪音(其中约 10-50% 的捕获率导致最低表达转录本的频繁丢失)与真正的生物噪音区分开来当蛋白质仍然存在时,转录物可能不会被转录并存在于细胞中一段时间。添加参考转录本和使用唯一分子标识符 (UMI) 已被应用于克服扩增偏倚和改善基因定量。可以量化基因水平技术变异的方法使我们能够专注于可能感兴趣的生物变异。典型的质量控制步骤包括留出包含很少读取的文库、具有低映射率的文库和具有零表达水平的看家基因(如 GAPDH 和 ACTB)的文库,这些基因预计以可检测的水平表达。根据所选的单细胞协议和实验目的,不同的批量 RNA-seq 管道和工具可用于不同阶段的分析。单细胞文库通常通过比对到参考转录组(使用 RSEM 等程序)进行分析,而无需尝试新的转录本发现,尽管存在一个软件可以比对到基因组(Monocle)。虽然比对到基因组确实会导致更高的整体reads比对率,但为了简单起见,仅关注基因表达且每个细胞读取较少的研究倾向于使用映射到参考转录组。已经开发了其他单细胞方法来使用 ATAC-seq测量单细胞 DNA 甲基化 和单细胞开放染色质。目前,我们只能在同一单个细胞中一次测量一种功能基因组数据类型,但我们可以预期,在不久的将来,我们将能够同时恢复单个细胞的转录组和其他功能数据。(一次放入样品,后面输出多种数据,转录组,基因组,等等)
(2)长度长测序(long-read sequencing)
短读长 RNA-seq 的主要限制是难以从读长的组装中准确重建表达的全长转录本。最初应用于基因组测序的长读长技术,例如 Pacific-Biosciences (PacBio) SMRT 和 Oxford Nanopore,现在正用于转录组学,并有可能克服这一组装问题。PacBio RNA-seq 是迄今为止发表最多的长读长方法。该技术已被证明可用于解开复杂基因的多种转录亚型,以及从单个读数中确定等位基因特异性表达 。
然而,长读长测序有其自身的一系列限制,例如仍然很高的错误率限制了从头转录本鉴定并迫使该技术利用参考基因组。此外,SMRT 细胞的相对低通量阻碍了转录表达的量化。
这两个限制可以通过将 PacBio 实验与常规的短读 RNA-seq 相匹配来解决。准确且丰富的 Illumina 读数可用于纠正长读长测序错误和量化转录水平。 PacBio 化学的更新正在增加测序长度,以产生具有足够数量的 cDNA 分子传递的读数,自动更正测序错误。这最终将提高测序准确性,并允许对异构体解析的转录组进行无基因组测定。
个人感悟
RNA-seq数据分析、解读和实际应用虽然常见,但其中诸多细节值得思考和学习,需要多多积累。关于其中RNA-seq的分析方法更是需要理解才行。
参考全文:
A survey of best practices for RNA-seq data analysis