StatQuest学习笔记23——RNA-seq简介
StatQuest学习笔记23——RNA-seq简介
前言——主要内容
这篇笔记是StatQuest系列笔记的第58节,主要内容是讲RNA-seq的原理。StatQuest系列教程的58到62节是协录组测序的内容。
RNA-seq研究的是什么
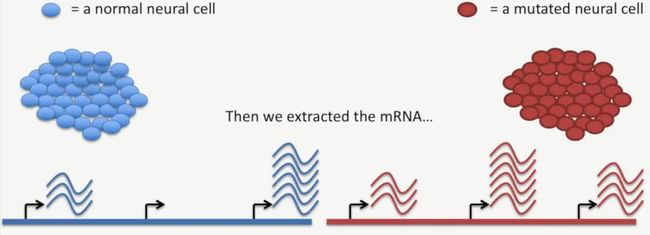
我们先来看一个案例,在下面的这个案例中,蓝色的细胞是一群正常的神经细胞,红色的细胞是一群突变的神经细胞。其中,突变的神经细胞表型与正常的神经细胞表型不同,此时,我们想知道,是什么遗传机制导致了这两群细胞表型的差异,这就意味着,我们要研究一下这两种细胞基因表达的差异,如下所示:

image
接下来,我们就来看一下,怎么找出这两群细胞基因表达的差异。
首先,根据高中的生物知识我就知道,一个细胞都有一群染色体(其数目因物种的不同而异),每条染色体上都有一些基因,在这些基因中,有些基因处于活跃状态,在下图中,这些活跃基因上面的波形图案表示这些基因mRNA的转录本,如下所示:

image
但是,还有一些基因是不活跃的,如下所示:

image
而高通量测序技术就能告诉我们,哪些基因是活跃的,以及它们的转录水平是多少,如下所示:
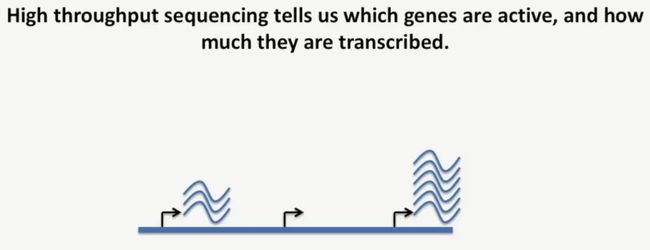
image
那么我们就可以通过RNA-Seq技术检测一下正常细胞的基因表达,再检测一下突变细胞的基因表达,如下所示:

image
然后我们比较一下这两种细胞基因表达的差异,如下所示:

image
例如,在这个案例中,我们就可以发现,经过RNA-Seq检测后,基因1的转录水平在这两种细胞内是没有差异的,如下所示:
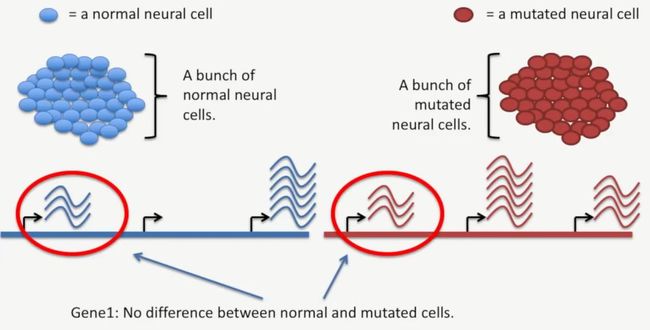
image
但是,基因2的转录则有很大的差异,如下所示:

image
基因3的转录水平也有差异,如下所示:
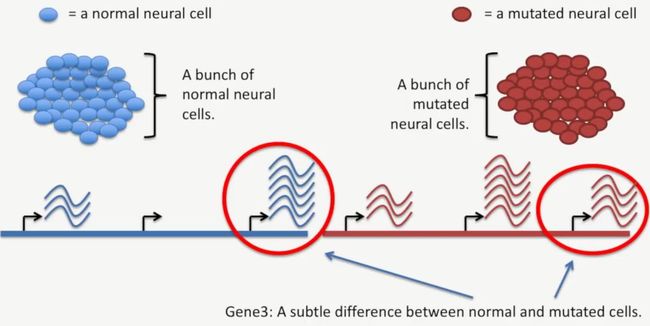
image
RNA-Seq的步骤
RNA-Seq主要有三个步骤,分别是第一:建库;第二,测序;第三,数据分析,如下所示:

image
第一步:建库
在这一步,我们就以Illumina的实验流程和测序仪为标准进行讲解,其他公司的流程和测序仪可能略有出入,如下所示:

image
建库又分这些步骤:
第一步,提取RNA;
第二步,将RNA打断成小的片段,打断的目的主要是因为RNA的长度有几千个碱基,而测序仪的读长只有200到300个bp,因此要进行打断,如下所示:
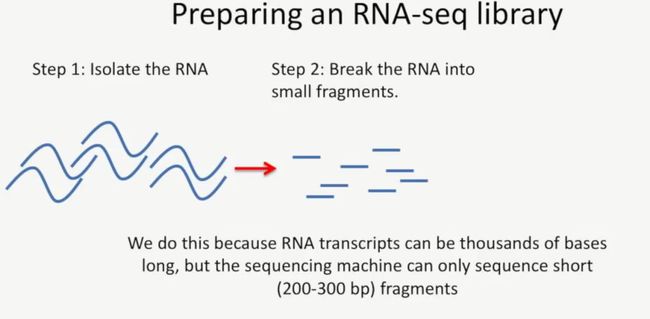
image
第三步,将RNA反转录为DNA,这一步的目的在于,双链DNA比RNA更加稳定,双链DNA更容易扩增与修饰,如下所示:

image
第四步,添加接头。接头主要发挥两个作用,第一,使测序仪识别加了接头的片段,因为接头上的序列与测序仪芯片上序列互补;第二,添加接头可以在一次测序中区分不同的样本,因为不同的样本可以使用不同的接头,如下所示:

image
但我们需要注意的是,在加接头这个步骤中,它的效率并不是100%的,有些片段并不会被加上接头,如下所示:

image
第五步:PCR扩增,这一步的扩增引物是接头上序列,只有那些加上了接头的序列才能扩增,如下所示:

image
第六步:质控。这一步主要是看两个指标:第一,确定文库的浓度,第二,确定文库的长度。确定文库的浓度方法就是(根据我们实验室自己的流程),用Qubit检测一下文库的浓度,这个浓度比较粗略,不能当成精确的数值,接着,使用qPCR的方法,对文库进行绝对定量,经过qPCR绝对定量方法得到的文库浓度才是最终的文库浓度。在确定文库片段的长度方面,通常是使用Agilent 2100进行检测,如下所示:
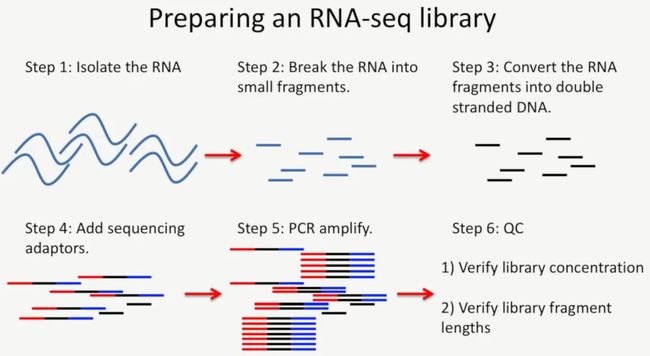
image
第二步:测序
测序测的其实就是文库,我们假设DNA片段的序列是下图的左侧部分,它是垂直的,因为在测序仪的芯片上,文库就是垂直排列的,在测序芯片上的一个小方格(grid)中将近有4亿条这样的序列,为了方便讲解,我们在下图的右侧只列出4条这样的序列,这样的一个小方格被称为flowcell,如下所示:

image
下图是一个flowcell:

image
在测序仪所使用的测序试剂中,含有一些特殊的碱基,这些碱基带有荧光探针,这些荧光探针按其结合碱基的不同,其颜色也不同,当测序仪开始测序时,这些带有荧光探针的碱基就会结合到DNA片段上第1个碱基,如下所示:

image
一旦这些带有荧光探针的碱基结合到DNA片段的碱基上,此时测序仪就会拍下一张照片,如果从上往下看,就是下图中红框所示的图片,这张图片会告诉测序仪,左下角的碱基是A,如下所示:

image
右下角的碱基就是G,如下所示:

image
左上角与右上角的碱基就是C,如下所示:

image
拍照结束后,测序仪会把这些这些碱基上的探针给冲走(测序试剂中有其他的成分,可以切掉这些荧光探针),此时,这些携带有探针的碱基就成了普通碱基,如下所示:
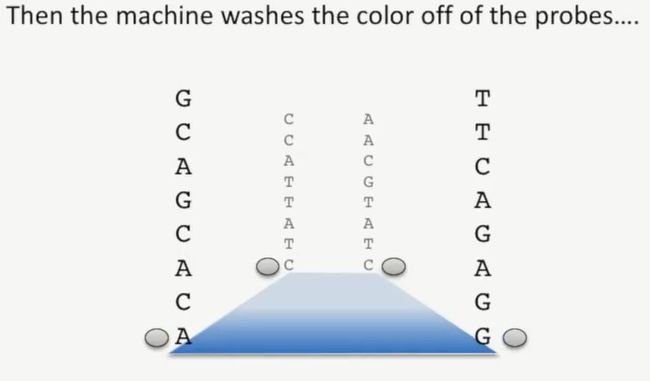
image
然后再加入含有荧光探针的碱基,再次与片段结合,如下所示:
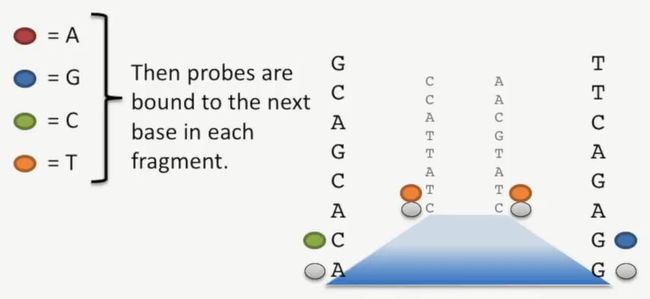
image
在第二次结合后,测序仪会拍照,从上往下看,就是下图红框内的图片,如下所示:

image
这样测序仪就知道,左下角的碱基是C,如下所示:

image
右下角的碱基是G,如下所示:

image
左上与右上的碱基是T,如下所示:
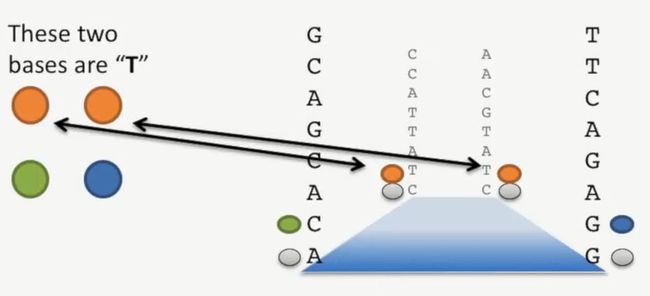
image
第2张图片识别后,再次用试剂切掉这些碱基上的荧光探针,并冲走,如下所示:
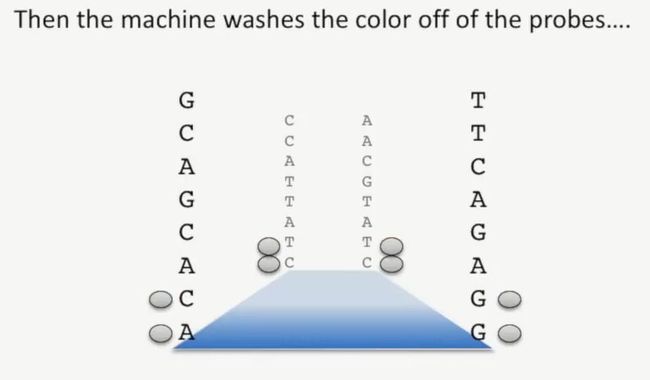
image
再进行第3次反应,如下所示:

image
总之,测序仪会不断地循环这个过程,直到测完所有的碱基,如下所示:

image
不过在实际的测序过程,这些DNA链的密度非常大,构成了一个密度极高的颜色矩阵,这个过程中也会产生一些问题,在下面我们就用一个简单的矩阵来说明这个问题,如下所示:

image
有时候,一个荧光探针的亮度可能不够,此时测序仪就没有足够的把握能够识别这种颜色,而在测序的过程中,根据这些探针的亮度,会生成一个质量评分(Quality score),这个质量评分反映了测序仪对这个颜色识别的可信程度,像在下面的这个图片中,这个比较暗的点可能就会得到一个比较低的质量评分,如下所示:

image
还有另外一种情况可能会得到一个质量评分,就是在某个区域内,相同的碱基数目太多,都呈现出一种颜色,如下图绿框所示部分,这种现象称为多样性差(low diversity),这种情况下,由于存在着大量的单一荧光,测序仪很难识别单独的碱基,这些颜色会混到一起。当测序仪测序时,对于文库中前几个碱基的识别很容易出现多样性差的问题,这是因为在刚开始的时候,测序仪要识别DNA片段位于芯片上的位置时,如下所示:

image
我们看一下测序后的原始数据,下图是测序的一个read的信息,它由4行构成,如下所示

image
第1行通常是由@开头的,它对于每条read,它都有唯一的ID,如下所示;

image
第2行是测序的文库片段的碱基序列,如下所示:

image
第3行是一个加号,它通常是空的,如下所示:
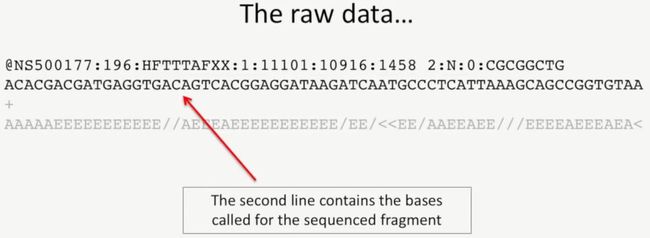
image
第4行是质量信息,它用一个字符表示这个字符对应的碱基的质量评分,如下所示:
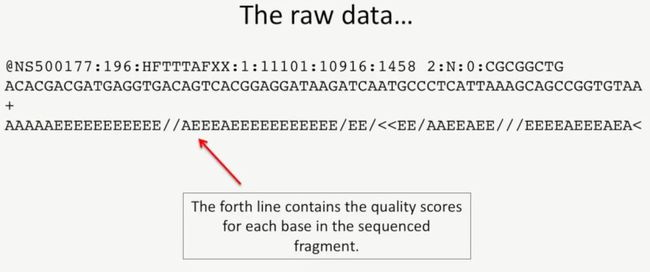
image
一次测序通常有4亿条reads数,一共会产生16亿行信息,如下所示:

image
数据预处理
我们现在已经知道了原始数据,以及原始数据如何产生的,那么我们下面要做这三件事情:
第一, 过滤掉垃圾reads;
第二,将高质量的reads比对到基因组上;
第三,对每个基因的reads数进行统计,如下所示:

image
过滤垃圾reads
垃圾reads是指:第一,某些reads的碱基质量低;第二,这些reads是明显的结合错误(第二种低质量的reads我不太清楚,原文我也看不太懂,我个人理解就是两个接头直接连接在一起的read)。
一条典型的read是一个DNA片段加上两个接头,但是在某些情况下,两个接头会直接加在一起,这就是垃圾reads,如下所示:

image
将read比对到基因组
我们先看一下基因组上的碱基序列,由于基因组的碱基序列很长,我们只截取一段出来,如下图中的红框所示,把这条基因组的碱基打断,会生成这些短的碱基序列,如下所示:

image
此时,我们把基因组的这些片段加上索引,并记录下它们在染色体上的位置,就是下图蓝框中的部分,如下所示:
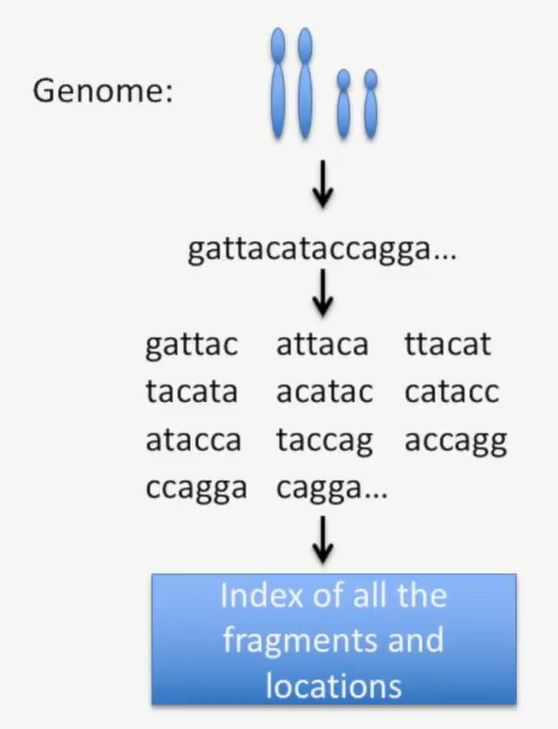
image
此时,我们把我们的测序read也打成小片段,就像上面的基因组打成小片段一样,如下所示:
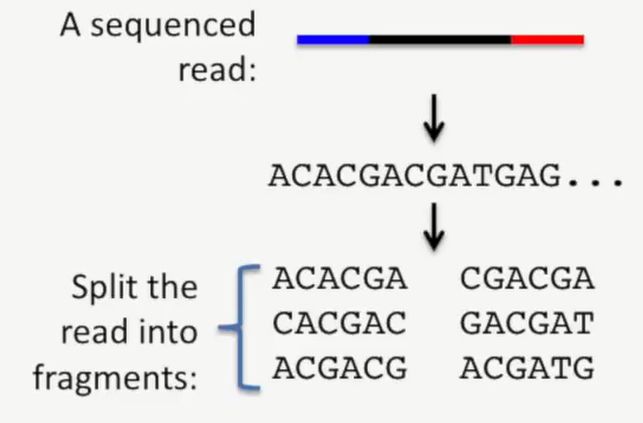
image
然后把reads的小片段与基因组的小片段进行匹配,如下所示:
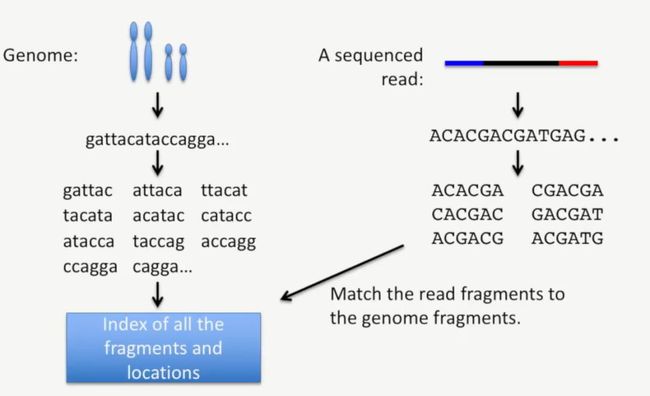
image
那些与reads的小片段匹配的基因组小片段就是这些read小片段在基因组上的位置(哪条染色体上,染色体的哪个位置),如下所示:

image
此时,我们可能有一个问题,为什么要把这些序列打断成小片段,这是因为即使reads与参考基因组在不是特别精确匹配的情况下,也能进行匹配,如下所示:
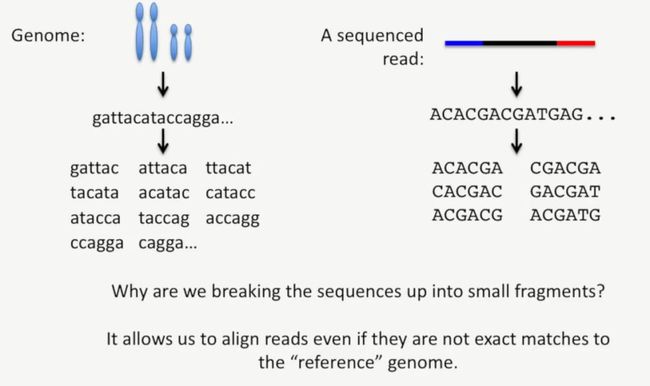
image
我们来看一个简单的例子,在下图中,某条read最左侧是A,而对应的基因组上并没有这个A(打个很简单的案例,我自己的基因组肯定与别人的基因组略有差异),如下所示:
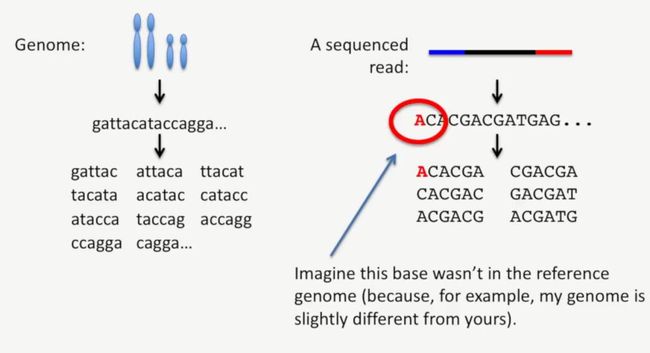
image
即使这个小片段无法与基因组上相匹配,那么其它的小片段还是能够匹配的,此时我们仍然可以找到这条read来源于基因组的哪个位置,如下所示:
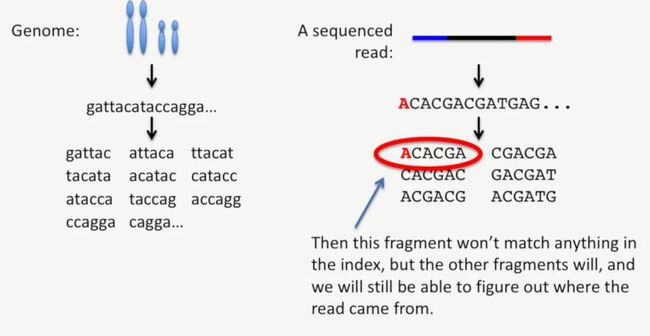
image
每个基因的reads计数
一旦我们知道了某条read的位置(也就是说知道了这条read在哪条染色体上,以及在染色体的哪个位置上),那么我们就可以看一下这条read是否能够落在某个基因的坐标中(或者是某些其它感兴趣的区域),例如在下图中,我们列出了Xkr4和Rp1这两个基因的坐标,如下所示:

image
当我们统计了每个基因的reads数后,我们就会得到下面这样的矩阵,如下所示:

image
第1列是基因名,在人类中,人类大概有20000个基因,因此这个矩阵的大概有2万行(下图中并没列完所有的基因),如下所示:
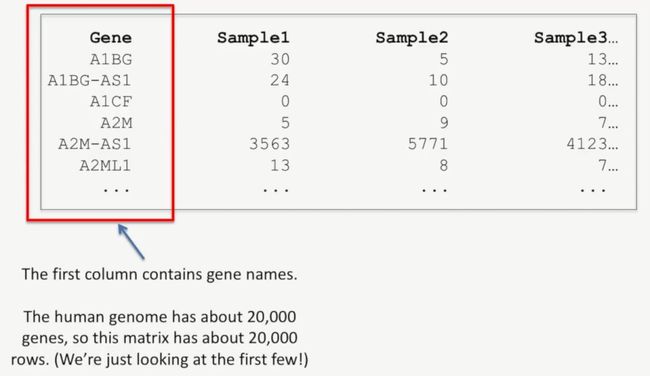
image
剩余的几列是每个样本对应的基因的reads数,这里的样本数目大概是6到800个,如下所示:

image
对于大部分的RNA-Seq来说,一个“样本”通常是一群细胞的平均值(通常是600万个细胞),一次实验,一般有3个正常的样本,3个疾病状态的样本,总共就是6个样本,如下所示:

image
而对于单细胞测序(Single-cell RNA-Seq)来说,每个细胞就是一个样本,因此这个矩阵中会出现有几百个样本,例如800多个,如下所示:

image
我们现在看某一行数据,如下所示,在这行数据中,我们可以看到每个样本中,每个特定基因的reads数,如下所示:

image
如果这个矩阵是单细胞测序的数据,那么这个矩阵有2万行(基因数目),800多列(样本数),大概有1600万个数值,这是一个极大的矩阵,并且随着测序技术发展,所测样本数目的增多,这样的矩阵会越来越大,如下所示:

image
在进行最终的数据分析之前,我们还要对数据进行均一化,这是因为每个样本比对到基因组上的reads数都不同,这可能是由于在测序时,有些样本的reads质量低,而另外某些样本的浓度略大,导致其总的reads数略高,如下所示:

image
例如,在下图中,Sample 1有635个reads比对上了,而Sample 2则有1270个reads数比对上了,Sample 2是Sample 1的两倍。但是这无法说明,Sample 2转录的基因是Sample 1的2倍,相反,这只能说明,Sample 2中的低质量reads数少,它在测序时,被测序仪识别的荧光更多而已,如下所示:

image
但是,我们发现,Sample 2中基因的reads数貌似是Sample 1中的基因reads数的2倍,如下所示:

image
A2M基因也是如此,如下所示:
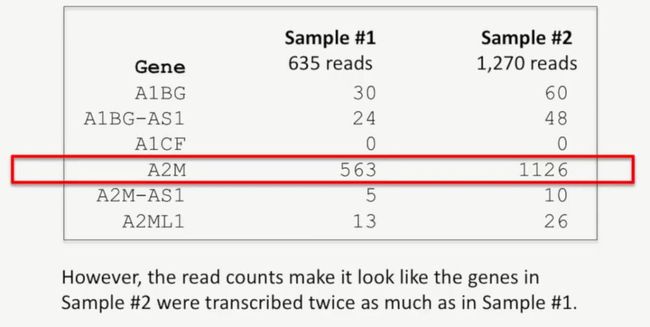
image
因此,我们需要调整每个基因的reads数,从这样才能真正反映出不同样本中比对上的reads数之间的差异,如下所示:

image
均一化最简单的方法就是在每个样本中,每个基因的reads数除以总的比对上的reads数,不过,还有其他更复杂的方法,如下所示:
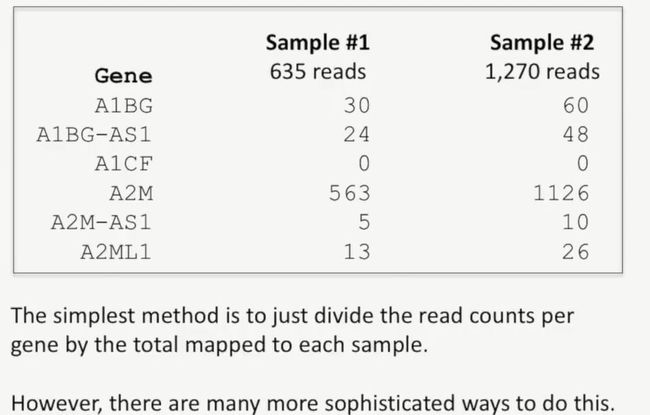
image
我们再回到最初的正常细胞与突变细胞的比较,我们先有了这两群细胞,如下所示:

image
然后提取RNA,如下所示:
image
随后,进行测序,比对,统计每个基因的reads数,然后均一化,如下所示:

image
此时,开始数据处理。
第三步:数据处理
数据处理的第一步通常都是相同,那就是绘图,我们要记住,这个表达矩阵非常大,如下所示:

image
如果每个样本只有2基因,那么绘图非常简单,如下所示:

image
第1步:绘图
首先我们用XY来替代这两个基因,根据它们的表达水平来绘图,如下所示:
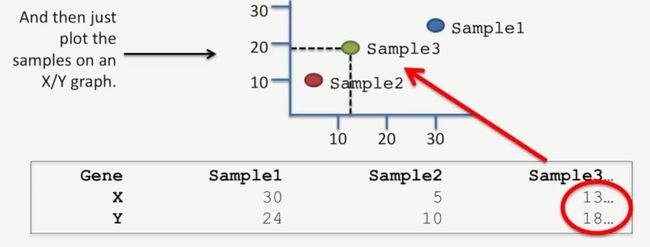
image
但是,我们有2万个基因,如下所示:

image
因此我们会用PCA或者是类似的方法来绘图,PCA能够降低坐标轴的数目(也就是把2万个基因对应的2万个坐标轴给降低到3个或2个),从而更加直观地表现数据,如下所示:

image
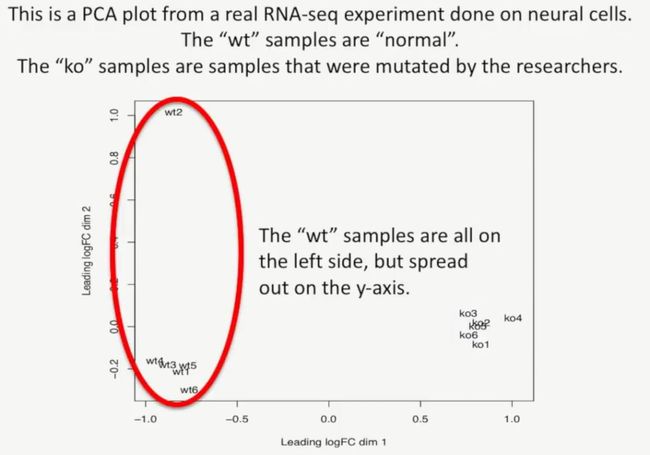
下图是我们利用PCA绘制的图形,正常的细胞是wt,突变的 细胞是ko,其中正常的细胞集中在左下角,敲除的细胞集中在右下方,如下所示:
image
从图中我们可以发现,这两类数据的差异主要体现在x轴上,如下所示:

image
相反,Y轴的差异没那么大,如下所示:

image
这就说明,wt组与ko组的差异主要集中于X轴,如下所示:

image
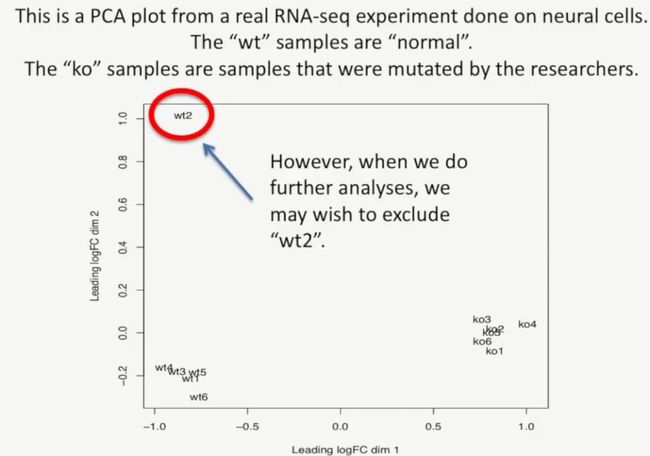
但是,当我们进行下一步的分析时,就需要排除wt2这个样本了,如下所示:
image
下图是单细胞测序的PCA图,如下所示:

image
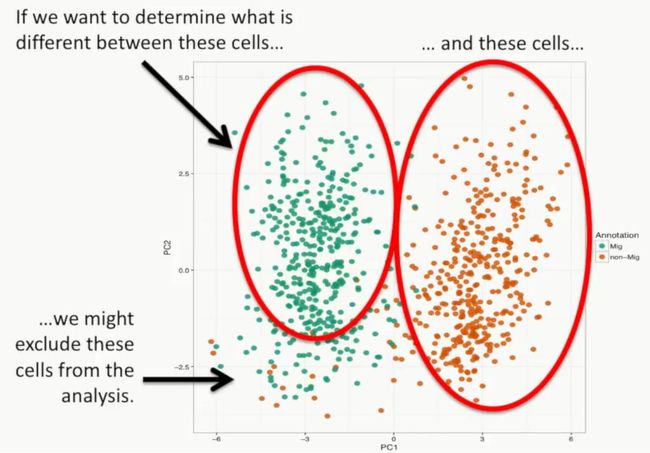
上面的不同颜色绘图依据是这些细胞行为的不同,绿色表示的是静止的细胞,橘黄色表示的是迁移到培养皿周围的细胞,如下所示:

image
大多数橘黄色的细胞与绿色的细胞是不同的,但是在左侧绿色的细胞中,也有几个是橘黄色的细胞,这说明这几个细胞的表型与绿色细胞更接近,如下所示:

image
如果我们想要研究上面两个大群细胞的差异,那么此时我们就要排除左下那几个橘黄色的细胞,如下所示:
image
总之,在对数据绘图后,我们可以从中得到这些信息:
- 找到感兴趣的差异部分;
- 在进行下游的分析之前,应该排除哪些数据,如下所示:

image
第2步:寻找差异基因
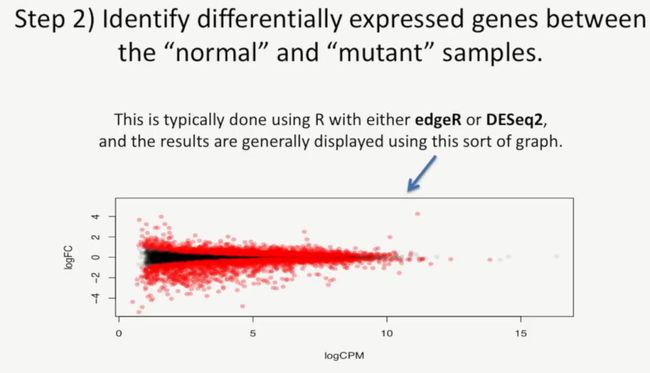
绘图后,我们就需要找到正常样本与突变样本有哪些差异表达基因,在分析差异基因时,通常使用R中的edgeR或DESeq2,它们通常以下图进行展示,如下所示:
image
其中红色部分是正常样本与突变样本的差异基因,如下所示:

image
中间的黑色部分是没有差异的基因,如下所示:

image
其中X轴表示的是基因的转录水平,它的单位是logCPM,其中CPM是counts per million的缩写,如下所示:

image
Y轴则是表示在正常样本与突变样本之间,差异基因的差异程度有多大,单位是logFC,即log(fold change),如下所示:

image
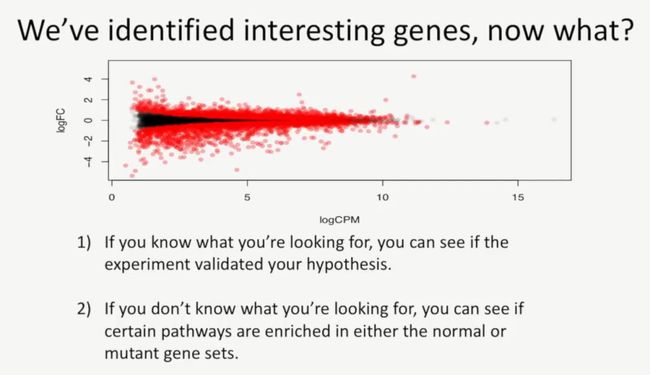
此时我们已经找到了感兴趣的基因(也就是差异基因),此时我们要做哪些事情呢?
第一,如果你知道你找的是什么(我的理解就是找的是具体的哪个基因),那么就要做实验,看能否验证你的假设;
第二,如果你不知道你接下来做什么,那么就你可以看一下这些差异基因集能否在某些通路上富集,如下所示:
image
每个样本对于每个特定的基因,它的reads数都不同。