扩增子分析流程 —— 数据处理(vsearch)
本篇文章是笔者学习了刘永鑫老师的扩增子教程以后,根据自己的学习过程总结了一篇使用vsearch工具处理序列数据得出OTU表的文章。笔者这篇文章会根据笔者自身的学习思路,每一步的数据处理都会从三个方面(是什么、为什么和怎么做)去分析展开。因为笔者以前没有接触过生信相关知识,所有生物知识还停留在高中水平,所以笔者所写的内容会比较偏向基础一些。如果本文章不符合你的需求,可以看刘永鑫老师的扩增子教程,不仅专业并且详细全面。
文章目录
-
- 扩增子测序简介
- 操作环境介绍
-
- 软件工具下载
-
- vsearch下载
- csvtk下载
- 第一步:获取样本元数据
-
- 操作名称
- 操作原因
- 操作方法
- 操作原理
- 第二步:获取测序数据
-
- 操作名称
- 操作原因
- 操作方法
- 第三步:获取数据库
-
- 操作名称
- 操作原因
- 操作方法
- 第四步:序列合并和重命名
-
- 操作名称
- 操作原因
- 操作方法
- 数据文件解读
- 第五步:切除引物与质控
-
- 操作名称
- 操作原因
- 操作方法
- 数据文件解读
- 第六步:序列去冗余
-
- 操作名称
- 操作原因
- 操作方法
- 数据文件解读
- 第七步:特征挑选
-
- 操作名称
- 操作原因
- 操作方法
- 数据文件解读
- 第八步:嵌合体检测
-
- 操作名称
- 操作原因
- 操作方法
- 第九步:生成特征表
-
- 操作名称
- 操作原因
- 操作方法
- 数据文件解读
- 第十步:物种注释
-
- 操作名称
- 操作原因
- 操作方法
- 数据文件解读
- 相关资源
- 参考文章
扩增子测序简介
扩增子测序是一种高靶向性方法(对特定长度的PCR产物或者捕获的片段进行测序),用于分析特定基因组区域中的基因变异。PCR产品(扩增子)的超深度测序可以有效地识别变异并对其进行特征分析。总体思路是靶向地捕获目标区域,然后进行新一代测序(NGS),分析测序结果数据,得到相应信息。
根据研究目的不同扩增子测序可细分为16S rDNA测序、18S rDNA测序、ITS测序及目标区域扩增子测序等。下面读者将以16S rDNA测序数据为例进行数据处理。
操作环境介绍
笔者这篇教程是在Ubuntu 20.04系统下完成的(如果读者使用的是win或mac系统,在处理过程中有配置错误的地方可以参考刘永鑫老师的教程,刘老师的教程很详细的说明了win和mac系统下的部署和处理过程)
笔者在完成这篇文章的时候新建了一个文件夹作为工作区,存放所有的处理过程文件,你可以根据你的习惯新建或者找一个文件夹作为工作区,这里要注意的是,笔者提供的所有命令都是在工作区下进行,如果你在子文件夹或者其他文件夹下需要修改路径。
软件工具下载
vsearch下载
-
vsearch工具下载
进入search下载页面进行选择你需要的版本进行下载,通常情况下是最新版本。
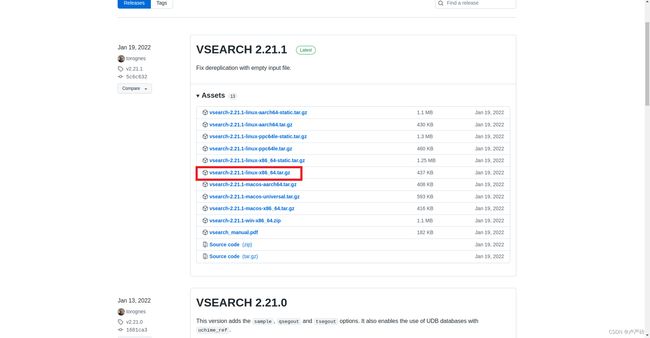
在工作区中新建文件夹“tools”,并且将下载的文件进行解压,并且将解压得到的文件夹打开,将bin文件夹下的vsearch文件复制到你新建的“tools”文件夹下。

-
配置环境变量
打开终端,输入以下命令:(如果使用的是无图形界面的系统,将gedit改为vim)
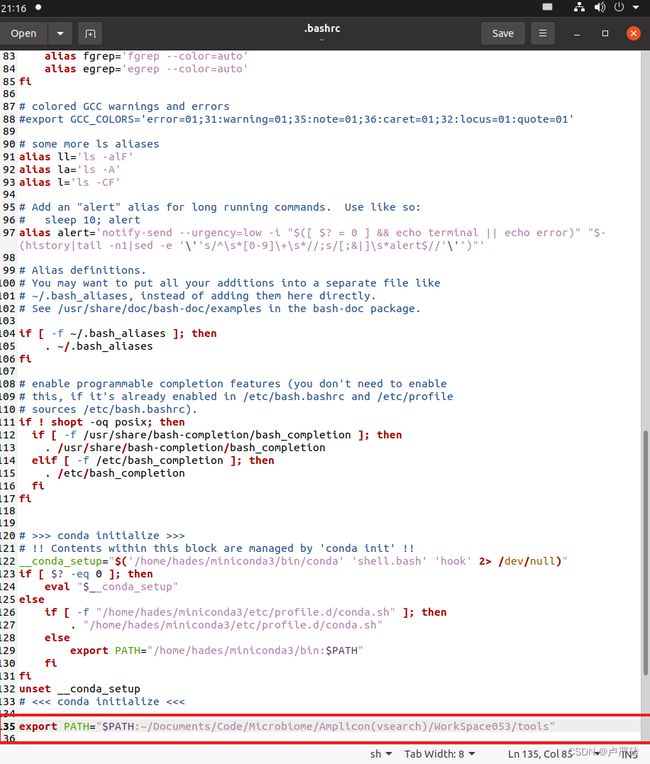
gedit ~/.bashrc在打开的文件中,底部添加
export PATH="$PATH:你tools文件夹的路径"并保存。例如笔者的路径为
~/Documents/Code/Microbiome/Amplicon(vsearch)/WorkSpace053/tools
-
测试是否安装成功
终端中继续输入以下命令,可以立即刷新修改的环境变量:
source ~/.bashrc然后在输入vsearch,显示以下内容,便是安装成功。

csvtk下载
-
该工具是用来方便在命令行内查看和处理“csv”格式文件的工具。
-
进入csvtk下载页面进行选择你需要的版本进行下载,通常情况下是最新版本。
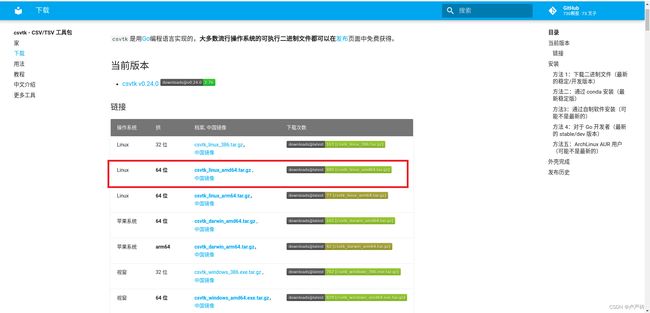
将下载的文件进行解压,并且将解压得到的文件夹打开,将文件夹下的csvtk文件复制到你新建的“tools”文件夹下。
-
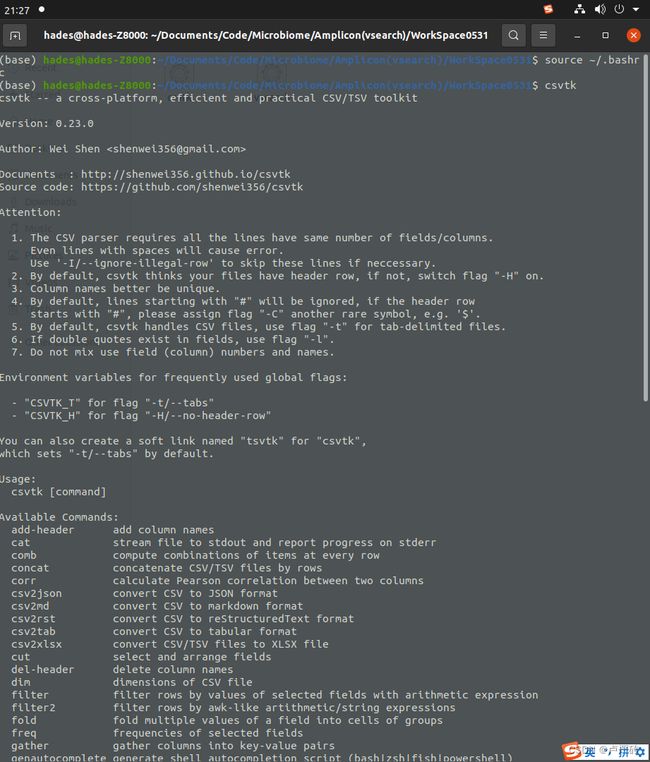
测试是否安装成功
终端中继续输入以下命令,立即刷新修改的环境变量:
source ~/.bashrc然后在输入csvtk,显示以下内容,便是安装成功。
-
第一步:获取样本元数据
操作名称
从各类高通量测序数据存放中心或者测序公司获取样本元数据
元数据(Metadata):又称中介数据、中继数据,为描述数据的数据(data about data),主要是描述数据属性(property)的信息,用来支持如指示存储位置、历史数据、资源查找、文件记录等功能。
样本元数据:在多次测序批次中用于记录每个样本的barcode、引物信息、序列研究中的分组、来自人类微生物组对应时间、时间点和身体部位等属性。
-
常见的高通量测序数据存放中心
- 国家基因组科学数据中心的GSA(Genome Sequence Archive)数据库
- NCBI的SRA(Sequence Read Archive)数据库
- EBI的ENA(European Nucleotide Archive)数据库
-
本片文章将介绍如何从我国建设的国家基因组科学数据中心获取元数据的方法。其他两个数据库的获取途径,笔者将在以后的文章中依次介绍。
操作原因
通过获取样本元数据不仅可以获取样本序列文件,还可以了解样本序列的各类属性信息。样本序列中的某些属性(比如:barcode、引物信息和分组信息等)会与后面的分析有关,所以必须要下载样本元数据来获取这些必要的属性信息。
操作方法
-
前往国家基因组科学数据中心的组学原始数据归档库;
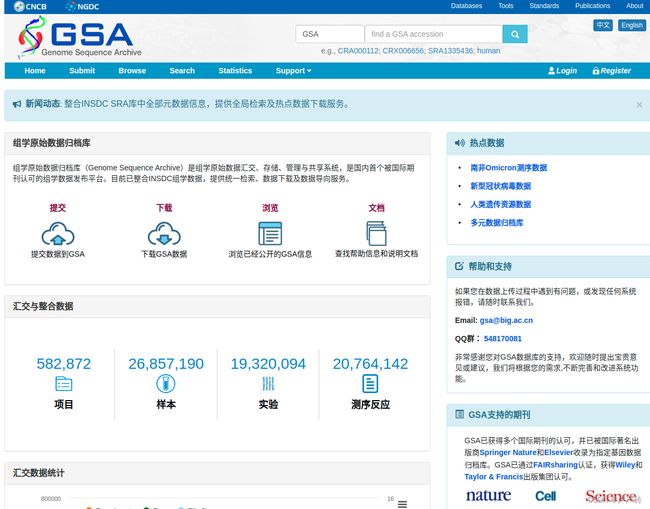
-
直接在网页顶部的搜索栏中输入序列号(accession)或者关键词,本例以序列号“CRA002352”和关键词“[A simple 16S amplicon project for pipeline test”为例进行查找,并点击页面的搜索(Search)按钮,跳转到搜索结果页面;
-
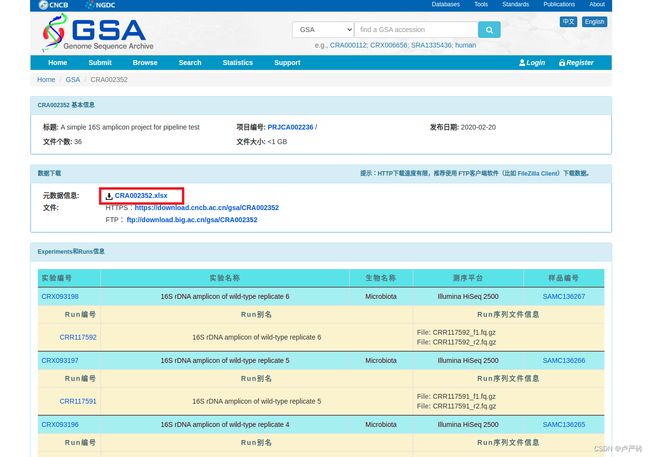
根据你的需求选择你所需要的样本数据,并点击你所需要的样本数据,跳转到样本数据的详细信息页面;
-
在详细页面的数据下载模块中,点击元数据信息旁的下载按钮,便可以获取样本元数据。

- 笔者为了方便数据的管理,讲序列数据都放在了“1_metadata”文件夹内,如果你想直接使用笔者后续的命令,这需要你提前在工作区也新建“1_metadata”文件夹,
mkdir -p 1_metadata。 - 当然你可以直接在本页面中通过https或者ftp方法直接下载序列数据,但也可以根据笔者后面写的第二步通过样本元数据的信息获取序列文件。
- 笔者为了方便数据的管理,讲序列数据都放在了“1_metadata”文件夹内,如果你想直接使用笔者后续的命令,这需要你提前在工作区也新建“1_metadata”文件夹,
-
样本元数据表格的解读
组学原始数据归档库中下载的样本元数据是excel格式的,主要有三个部分,所以样本元数据表格有3个工作表,分别是样本(Sample)、实验(Experiment)和测序反应(Run)。如下是三个工作表的展示和每列内容的描述:
VSCode + Excel Viewer: 笔者在整个扩增子分析流程中,都是在Ubuntu20.04系统环境下完成的,在Ubuntu系统中查看Excel表格文件笔者推荐用VSCode,并下载“Excel Viewer”插件。
-
样本表(Sample)
ID Sample name Accession Public description Project accession Sample title Organism Host Isolation source Collection date Geographic location Latitude longitude Reference biomaterial Relationship to oxygen Sample collection device Sample material processing Sample size Source material identifiers Description 1 KO1 SAMC136250 Knock-out replicate 1 PRJCA002236 KO1 Microbiota Arabidopsis thaliana Arabidopsis root 2017-06-30 China: Beijing 40.00 N 116.22 E Col-0 30days 2 KO2 SAMC136251 Knock-out replicate 2 PRJCA002236 KO2 Microbiota Arabidopsis thaliana Arabidopsis root 2017-06-30 China: Beijing 40.00 N 116.22 E Col-0 30days 3 KO3 SAMC136252 Knock-out replicate 3 PRJCA002236 KO3 Microbiota Arabidopsis thaliana Arabidopsis root 2017-07-02 China: Beijing 40.00 N 116.22 E Col-0 30days 4 KO4 SAMC136253 Knock-out replicate 4 PRJCA002236 KO4 Microbiota Arabidopsis thaliana Arabidopsis root 2017-07-02 China: Beijing 40.00 N 116.22 E Col-0 30days 列名 描述信息 ID 索引(编号必须唯一) Sample name 样本名称(与实验和测序反应模块相对应) Accession 样本序列号 Public description 样本的描述信息 Project accession 项目序列号 Sample title 样本标题 Organism 组织来源的物种学名,即拉丁名称 Host 样本来源生物的天然(非实验室)宿主物种学名,即拉丁名称 Isolation source 分离提取生物样本的物理、环境和/或地理分部信息 Collection date 采样时间 Geographic location 采样地点 Latitude longitude 采样地点的地理坐标 Reference biomaterial 生物材料参考文章出处或基因组报告出处 Relationship to oxygen 微生物生存与氧气的关系(好氧、需氧、耐氧、厌氧等) Sample collection device 采集样本的方法和装置 Sample meterial processing 分离期间或分离后对样本的处理 Sample size 收集的样本的数量或大小 Source material identifiers 用于提取核酸和测序的样本材料的唯一标识符 Description 自定义补充信息 -
实验表(Experiment)
ID Accession Experiment title BioProject accession BioSample name BioSample accession Platform Library Construction / Experimental Design Library name Strategy Source Selection Layout Read length for mate1(bp) Read length for mate 2(bp) Insert size (bp) Nominal size (bp) Nominal standard deviation (bp) Planned number of cycles other_db accession_in_other_db other_db_url 1 CRX093181 16S rDNA amplicon of knock-out replicate 1 PRJCA002236 KO1 SAMC136250 Illumina HiSeq 2500 DNA for each sample was extracted ······ AMPLICON METAGENOMIC PCR PAIRED 250 250 441 2 CRX093182 16S rDNA amplicon of knock-out replicate 2 PRJCA002236 KO2 SAMC136251 Illumina HiSeq 2500 DNA for each sample was extracted ······ AMPLICON METAGENOMIC PCR PAIRED 250 250 441 3 CRX093183 16S rDNA amplicon of knock-out replicate 3 PRJCA002236 KO3 SAMC136252 Illumina HiSeq 2500 DNA for each sample was extracted ······ AMPLICON METAGENOMIC PCR PAIRED 250 250 441 4 CRX093184 16S rDNA amplicon of knock-out replicate 4 PRJCA002236 KO4 SAMC136253 Illumina HiSeq 2500 DNA for each sample was extracted ······ AMPLICON METAGENOMIC PCR PAIRED 250 250 441 列名 描述信息 ID 索引(编号必须唯一) Experiment title 实验标题 BioProject accession 项目序列号 BioSample name 样本名称样本名称(与实验和测序反应模块相对应) BioSample accession 样本序列号 Platform 测序平台 Library Construction / Experimental Design 简介你的测序前实验,如DNA提交、扩增引物、建库方法等 Library name 文库名称,样本来源的文库ID,可用于研究批次效应 Strategy 建库类型 Source 实验材料来源类型 Selection 片段的富集或选择方法 Layout 测序模式 Read length for mate1(bp) 正向测序的长度 Read length for mate2(bp) 反向测序的长度 Insert size (bp) 测序片段长度 Nominal size (bp) 测序片段的标准长度 Nominal standard deviation (bp) 标准差 Planned number of cycles 计划周期数 other_db 是否存在其他数据库 accession_in_other_db 其他数据库的序列号 other_db_url 其他数据库的链接 -
测序反应表(Run)
ID Accession Run title BioProject accession Experiment accession Run data file type File name 1 MD5 checksum 1 DownLoad1 File name 2 MD5 checksum 2 DownLoad2 Reference file name MD5 for reference file Assembly Name or Accession Assembly Accession URL other_db accession_in_other_db other_db_url 1 CRR117575 16S rDNA amplicon of ······ PRJCA002236 CRX093181 fastq CRR117575_f1.fq.gz (6516124 bytes) 180d95da80536083bab9f5059e9d300c ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz CRR117575_r2.fq.gz (7131045 bytes) 33756be503f150603ef2aa4808aa3016 ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_r2.fq.gz 2 CRR117576 16S rDNA amplicon of ······ PRJCA002236 CRX093182 fastq CRR117576_f1.fq.gz (6556819 bytes) c8bd3361ce2d3e192bc235540fffa995 ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_f1.fq.gz CRR117576_r2.fq.gz (7115422 bytes) 1edb49a24f10decc6101feb0c5bf9ac1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_r2.fq.gz 3 CRR117577 16S rDNA amplicon of ······ PRJCA002236 CRX093183 fastq CRR117577_f1.fq.gz (6434960 bytes) 7617114a1f4a64c04bae83208e07d21c ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_f1.fq.gz CRR117577_r2.fq.gz (7020662 bytes) 7c3d4571c13a1d0347722e69ed899628 ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_r2.fq.gz 4 CRR117578 16S rDNA amplicon of ······ PRJCA002236 CRX093184 fastq CRR117578_f1.fq.gz (6395711 bytes) 1d98eb7814fcd509bfa61e44b76ba823 ftp://download.big.ac.cn/gsa/CRA002352/CRR117578/CRR117578_f1.fq.gz CRR117578_r2.fq.gz (7036983 bytes) f80dc414720a3ef9e41af1c5a11c739a ftp://download.big.ac.cn/gsa/CRA002352/CRR117578/CRR117578_r2.fq.gz 列名 描述信息 ID 索引(编号必须唯一) Run title 测序反应标题 BioProject accession 项目序列号 Experiment accession 环境序列号 Run data file type 实验数据文件类型 File name 1 测序文件1名称 MD5 checksum 1 测序文件1的MD5检验码 Download1 测序文件1的下载链接 File name 2 测序文件2名称 MD5 checksum 2 测序文件2的MD5检验码 Download2 测序文件2的下载链接 Reference file name 参考文件名称 MD5 for reference file 参考文件的MD5校验码 Assembly Name or Accession 参考序列名称或序列号 Assembly Accession URL 参考序列对应的网址链接 other_db 是否存在其他数据库 accession_in_other_db 其他数据库的序列号 other_db_url 其他数据库的链接
-
操作原理
- 国家的高通量测序数据中心是通过将按照规范上传的组学原始数据建立成的基因组序列存档数据库,多以我们在上传时就必须填写测序数据的样本元数据,上传成功后生成上传项目序列号。这样我们才能够更具序列号或者关键词下载已经上传好的样本元数据,然后后续再根据样本元数据的属性信息下载序列数据。
- 这里需要提到的是在发表论文等操作时,是需要将论文中涉及到的序列信息,按照数据中心的格式上传到相应的数据中心的,所以了解样本元数据中各属性代表的信息还是很有必要的。
第二步:获取测序数据
操作名称
根据样本元数据获取测序数据
测序数据:通常为一个样品一对fq/fastq.gz格式压缩文件(如果是单端测序则只有一个)
测序数据文件格式:
数据库文件是“.fq”后缀,该类型的文件格式是“fastq”。
“fastq”格式的文件中每个序列主要有四个部分组成:
第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;
第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。
其中碱基的质量如何评估在本篇文章中不详细展开,需要的了解的可以去usearch官网文档的这篇文章中查看。
操作原因
必须要获取了测序数据才能够对扩增子测序序列进行分析
操作方法
-
打开第一步获取的样本元数据表格,切换到测序反应(Run)的工作表;
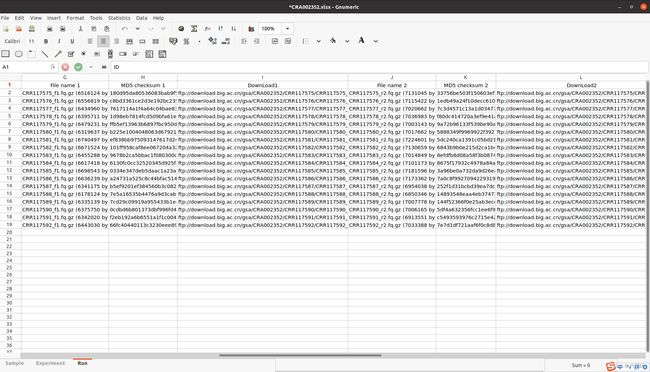
-
根据列“DownLoad1”和“DownLoad2”中的文件链接,进行下载;
-
逐个下载
如果序列数据较少,可根据样本元数据中提供的ftp链接,在Ubuntu系统中使用wget命令进行下载,只需要逐个替换以下命令行的ftp链接便可以完成所有序列下载。
wget -c ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz -O seq/KO1_1.fq.gz- wget: wget命令是Linux系统用于从Web下载文件的命令行工具,支持 HTTP、HTTPS及FTP协议下载文件。 -c 参数是断点续传的命令, -O 参数是用来将文件下载到指定路径和文件名;
- 笔者为了方便数据的管理,将序列数据都放在了“2_seq”文件夹内,这需要你提前在工作区也新建“2_seq”文件夹,
mkdir -p 2_seq。

-
批量下载(稍有难度,需要对linux的命令行工具有所了解)
如果样本数据较多,可以设计相应的命令语句来执行批量操作,但是涉及到比较多的命令行工具,以下是笔者的涉及思路,如有不理解可以联系笔者。
-
获取所有的下载链接和样本名称到csv格式文件*(因为官网提供的样本元数据格式是excel文件,使用命令行语句批量不容易操作,所以思路是将数据提取到容易操作的文件格式中,例如:csv、txt)*
-
方法一:
直接将样本元数据中的下载数据和样本数据手动粘贴到新建的csv格式文件。本篇文章笔者命名为“metadata.csv”
KO1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_r2.fq.gz KO2 ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_r2.fq.gz KO3 ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_r2.fq.gz KO4 ftp://download.big.ac.cn/gsa/CRA002352/CRR117578/CRR117578_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117578/CRR117578_r2.fq.gz KO5 ftp://download.big.ac.cn/gsa/CRA002352/CRR117579/CRR117579_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117579/CRR117579_r2.fq.gz KO6 ftp://download.big.ac.cn/gsa/CRA002352/CRR117580/CRR117580_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117580/CRR117580_r2.fq.gz OE1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117581/CRR117581_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117581/CRR117581_r2.fq.gz OE2 ftp://download.big.ac.cn/gsa/CRA002352/CRR117582/CRR117582_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117582/CRR117582_r2.fq.gz OE3 ftp://download.big.ac.cn/gsa/CRA002352/CRR117583/CRR117583_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117583/CRR117583_r2.fq.gz OE4 ftp://download.big.ac.cn/gsa/CRA002352/CRR117584/CRR117584_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117584/CRR117584_r2.fq.gz OE5 ftp://download.big.ac.cn/gsa/CRA002352/CRR117585/CRR117585_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117585/CRR117585_r2.fq.gz OE6 ftp://download.big.ac.cn/gsa/CRA002352/CRR117586/CRR117586_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117586/CRR117586_r2.fq.gz WT1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117587/CRR117587_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117587/CRR117587_r2.fq.gz WT2 ftp://download.big.ac.cn/gsa/CRA002352/CRR117588/CRR117588_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117588/CRR117588_r2.fq.gz WT3 ftp://download.big.ac.cn/gsa/CRA002352/CRR117589/CRR117589_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117589/CRR117589_r2.fq.gz WT4 ftp://download.big.ac.cn/gsa/CRA002352/CRR117590/CRR117590_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117590/CRR117590_r2.fq.gz WT5 ftp://download.big.ac.cn/gsa/CRA002352/CRR117591/CRR117591_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117591/CRR117591_r2.fq.gz WT6 ftp://download.big.ac.cn/gsa/CRA002352/CRR117592/CRR117592_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117592/CRR117592_r2.fq.gz -
方法二:
先将样本元数据的文件格式转为csv格式
sudo apt-get install gnumeric在Linux上,将xlsx Excel格式文件转换为CSV,需要使用Gnumeric电子表格程序
ssconvert -S 1_metadata/CRA002352.xlsx 1_metadata/CRA002352.csv可以看到样本元数据表的三个sheet导出为三个csv文件。样本表(CRA002352.csv.1)、实验表(CRA002352.csv.1)和测序反应表(CRA002352.csv.2
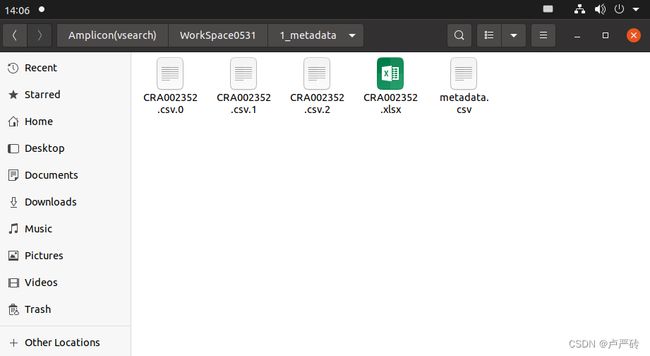
ssconvert:ssconvert是Gnumeric电子表格程序中的一个命令行工具,可以将“xls”、“xlsx”、"csv"等格式文件之间互相转化。-S 参数是将表格每个的sheet都导出为一个csv文件
然后将下载链接和样本名称通过命令行语句提取出,并导入到新的csv格式文件中。
首先是将存有样本名称的实验表(CRA002352.csv.1)和测序反应表(CRA002352.csv.2)通过关键列进行合并。本例中,实验表的关键列是“Accession”,测序反应表的关键列是“Experiment accession”。合并以后的数据表如下。
ID Accession Experiment title BioProject accession BioSample name BioSample accession Platform Library Construction / Experimental Design Library name Strategy Source Selection Layout Read length for mate1(bp) Read length for mate 2(bp) Insert size (bp) Nominal size (bp) Nominal standard deviation (bp) Planned number of cycles other_db accession_in_other_db other_db_url ID Accession Run title BioProject accession Run data file type File name 1 MD5 checksum 1 DownLoad1 File name 2 MD5 checksum 2 DownLoad2 Reference file name MD5 for reference file Assembly Name or Accession Assembly Accession URL other_db accession_in_other_db other_db_url 1 CRX093181 16S rDNA amplicon ··· PRJCA002236 KO1 SAMC136250 Illumina HiSeq 2500 DNA for each sample was ··· AMPLICON METAGENOMIC PCR PAIRED 250 250 441 1 CRR117575 16S rDNA ··· PRJCA002236 fastq CRR117575_f1.fq.gz (6516124 bytes) 180d95da80536083bab9f5059e9d300c ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz CRR117575_r2.fq.gz (7131045 bytes) 33756be503f150603ef2aa4808aa3016 ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_r2.fq.gz 2 CRX093182 16S rDNA amplicon ··· PRJCA002236 KO2 SAMC136251 Illumina HiSeq 2500 DNA for each sample was ··· AMPLICON METAGENOMIC PCR PAIRED 250 250 441 2 CRR117576 16S rDNA ··· PRJCA002236 fastq CRR117576_f1.fq.gz (6556819 bytes) c8bd3361ce2d3e192bc235540fffa995 ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_f1.fq.gz CRR117576_r2.fq.gz (7115422 bytes) 1edb49a24f10decc6101feb0c5bf9ac1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_r2.fq.gz 3 CRX093183 16S rDNA amplicon ··· PRJCA002236 KO3 SAMC136252 Illumina HiSeq 2500 DNA for each sample was ··· AMPLICON METAGENOMIC PCR PAIRED 250 250 441 3 CRR117577 16S rDNA ··· PRJCA002236 fastq CRR117577_f1.fq.gz (6434960 bytes) 7617114a1f4a64c04bae83208e07d21c ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_f1.fq.gz CRR117577_r2.fq.gz (7020662 bytes) 7c3d4571c13a1d0347722e69ed899628 ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_r2.fq.gz 发现样本名称列在第5列,第一个序列文件的下载链接在第30列,第二个序列文件的下载链接在第33列。于是提取出这三列数据通过重定向命令新建csv格式文件,将下载链接和样本名称保存在里面。
csvtk join -f 'Accession;Experiment accession' 1_metadata/CRA002352.csv.1 1_metadata/CRA002352.csv.2 | tail -n +2 | csvtk cut -f 5,30,33 > 1_metadata/metadata.csvcsvtk:
- join -f <关键词> <文件名 文件名> 参数是用来通过关键词将两个csv文件合并在一起,本文的关键词是 ‘Accession;Experiment accession’,合并文件为*“1_metadata/CRA002352.csv.1 1_metadata/CRA002352.csv.2”*;
- cut -f <行数,行数> 参数是挑选特定列的数据,本文是挑选第5列和第30/33列的数据。
tail:tail 命令可用于查看文件的内容。-n <行数> 参数是显示文件的尾部 n 行内容,但是如果参数前有正好(+n)则表示从第n行至文件末尾;
整个的命令语句是先用csvtk工具合并转化后的表格1和表格2,然后tail工具获取去表头的数据 (从第2行至文件末尾),然后通过管道命令(|)将数据传递给csvtk工具获取样本名称和下载列的ftp链接数据 (笔者的是第5列和第30和33列的数据,读者根据自己情况进行修改),然后将读取的样本名称和ftp链接数据重定向(>)给命名为“metadata.csv”的文件,并保存。
KO1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_r2.fq.gz KO2 ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117576/CRR117576_r2.fq.gz KO3 ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117577/CRR117577_r2.fq.gz KO4 ftp://download.big.ac.cn/gsa/CRA002352/CRR117578/CRR117578_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117578/CRR117578_r2.fq.gz KO5 ftp://download.big.ac.cn/gsa/CRA002352/CRR117579/CRR117579_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117579/CRR117579_r2.fq.gz KO6 ftp://download.big.ac.cn/gsa/CRA002352/CRR117580/CRR117580_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117580/CRR117580_r2.fq.gz OE1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117581/CRR117581_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117581/CRR117581_r2.fq.gz OE2 ftp://download.big.ac.cn/gsa/CRA002352/CRR117582/CRR117582_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117582/CRR117582_r2.fq.gz OE3 ftp://download.big.ac.cn/gsa/CRA002352/CRR117583/CRR117583_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117583/CRR117583_r2.fq.gz OE4 ftp://download.big.ac.cn/gsa/CRA002352/CRR117584/CRR117584_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117584/CRR117584_r2.fq.gz OE5 ftp://download.big.ac.cn/gsa/CRA002352/CRR117585/CRR117585_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117585/CRR117585_r2.fq.gz OE6 ftp://download.big.ac.cn/gsa/CRA002352/CRR117586/CRR117586_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117586/CRR117586_r2.fq.gz WT1 ftp://download.big.ac.cn/gsa/CRA002352/CRR117587/CRR117587_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117587/CRR117587_r2.fq.gz WT2 ftp://download.big.ac.cn/gsa/CRA002352/CRR117588/CRR117588_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117588/CRR117588_r2.fq.gz WT3 ftp://download.big.ac.cn/gsa/CRA002352/CRR117589/CRR117589_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117589/CRR117589_r2.fq.gz WT4 ftp://download.big.ac.cn/gsa/CRA002352/CRR117590/CRR117590_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117590/CRR117590_r2.fq.gz WT5 ftp://download.big.ac.cn/gsa/CRA002352/CRR117591/CRR117591_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117591/CRR117591_r2.fq.gz WT6 ftp://download.big.ac.cn/gsa/CRA002352/CRR117592/CRR117592_f1.fq.gz ftp://download.big.ac.cn/gsa/CRA002352/CRR117592/CRR117592_r2.fq.gz
-
-
进行批量下载
# 批量下载序列文件1并重命名 awk '{system("wget -c "$2" -O 2_seq/"$1"_1.fq.gz")}' <(cat 1_metadata/metadata.csv | csvtk cut -f 1,2 | csvtk csv2tab) # 批量下载序列文件2并重命名 awk '{system("wget -c "$2" -O 2_seq/"$1"_2.fq.gz")}' <(cat 1_metadata/metadata.csv | csvtk cut -f 1,3 | csvtk csv2tab)awk: awk是一个优良的文本处理工具,Linux及Unix环境中现有的功能最强大的数据处理引擎之一。
- awk处理脚本的格式为
awk '{awk脚本}'; - awk脚本中的system()函数是可以直接在awk中调用shell命令,会启动一个新shell进程执行命令,需要处理的命令就是wget下载命令,其中**“$1”是用来获取重定向命令**(<)传递的参数;
cat:cat命令可用于查看文件的内容;
csvtk:
- cut -f <行数,行数> 参数是挑选特定列的数据,本文是挑选第5列和第30/33列的数据;
- csv2tab 参数是将csv表格输出为制表符分割格式(因为awk工具如果有多个输入参数,需要空格分开);
wget:-O 参数是指定文件名;
整个的命令语句是先用cat命令查看文件内容,然后通过管道命令(|)将数据传递给csvtk工具获取样本名称和下载列的ftp链接数据 (分别是第1列和第2/3列),然后将读取的样本名称和ftp链接数据重定向(<)给awk工具进行批量脚本处理。
- awk处理脚本的格式为
获取到的测序数据文件共36个文件。
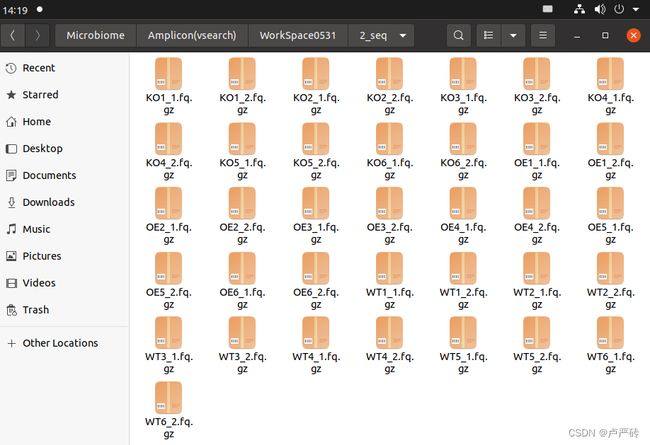
-
-
第三步:获取数据库
操作名称
-
通过公共数据库中心获取扩增子的参考数据库
扩增子参考数据库:主要收录微生物的16S,18S个ITS序列信息。
数据库种类:
-
GreenGenes:
GreenGene是一个最经典的16S物种数据库,专门针对细菌、古菌16S rRNA基因。该数据库是基于人工整理,数据更加比较准确。分类上采用的是通用的界门纲目科属种七级,方便使用者理解和阅读。可惜的是,该数据库很久未更新了,目前的最新版本还是2013年更新的。但是很多实验室还是热衷于用该数据库进行注释,而且PICRUST、QIIME等诸多工具也是基于该数据库设计的。
-
SILVA:
SILVA数据库是收纳细菌、古菌和真核微生物的rRNA基因序列的综合数据库,该数据库包含了原核和真核微生物的小亚基rRNA(16S和18SrRNA)和大亚基rRNA(23S和28SrRNA)序列。该数据库更新很频繁,但是数据假阳性较greengene高。而且该数据库的物种注释采用的是14级,与常用的七级不同,不方便比较。
-
RDP:
RDP数据库的全称是“RibosomalDatabase Project”。是由密歇根州立大学开发维护的在线工具,内容主要包括了数据库和在线分析工具2部分。其中,数据库部分提供了细菌、古菌的16S rRNA基因以及真菌28S rRNA基因序列。其最新版本是2016年9月更新,包含了3356809条16S rRNA基因序列和125525条真菌28S rRNA基因序列。
-
UNITE:
UNITE数据库是专门针对真菌ITS序列,包括ITS1和ITS2区最全面的数据库。ITS是最常用的真菌鉴定及多样性检测的marker基因,UNITE是ITS高通量测序后对真菌进行分类注释的比对最常用的数据库。该数据库最新版本是2017年12月更新,新版本包含 817130条ITS序列,可以下载数据库,也可以在线对ITS序列进行鉴定(https://unite.ut.ee/analysis.php)。
-
PR2:
PR2数据库是专门针对真核微生物18SrRNA基因的数据库。该数据库主要由核编码的原生生物序列构成,但为方便分析18S的高通量测序数据,数据库也包含了后生生物、陆地植物、大型真菌和真核细胞器(线粒体、质体等)的SSU序列。该数据最新更新是在2018年2月。
-
FunGene:
FunGene是一个针对微生物功能基因序列的数据库,而且提供了一些工具对功能基因进行分析。FunGene数据库将功能基因分为了7类,抗生素抗性(Antibiotic resistances)、生物地球化学循环(Biogeochemical cycles)、植物的致病基因(Plant Pathogenicity)、系统进化标记(Phylogenetic markers)、生物降解(Biodegradation)、金属循环(Metal Cycling)和其他(Other。FunGene的序列来源于GeneBank 数据库,而GeneBank 数据库是有冗余的,所以FunGene 也会有冗余现象,所以在下载完序列之后,需要去冗余。FunGene可被用于功能marker基因高通量测序后的比对以及功能基因的引物设计等。
-
NCBI
研究中有时不只有细菌、真菌;比如18S/ITS可以扩增出所有真核生物,最全的数据库那当然还是NCBI。很可惜这么强大的需求下没有整理。想要注释最全的物种信息,大家只能按文中的说明,自己整理吧。注:NCBI数据无人把关,比较乱,假阳性率或错误比较多;但有信息总比没有强。
通常注释需要将序列blast到NCBI的NR的核酸或蛋白库中,获得最相拟序列,再结果相似序列的GI号转换Taxonomy,链接中有详细的教程。
数据库文件格式:
- 数据库文件是“.fa”后缀,该类型的文件格式是“fasta”。
- “fasta”格式的文件中每个序列主要有两个部分组成:
- 序列头信息(有时包括一些其它的描述信息)和具体的序列数据。
- 序列内容
- “fasta”格式的文件与“fastq”格式文件最大的差别是,“fasta”格式文件没有第三行和第四行的序列质量值。
-
操作原因
- 扩增子测序数据分析最主要的一个材料就是扩增子数据库,用来与测序数据进行比对,并进行物种注释;
- 在分析流程后期中基于参考数据库去嵌合和去质体和非细菌等步骤中也需要用到扩增子数据库。
操作方法
-
关于vsearch工具中用到的数据库,最常见的途径是去usearchs官网获取;
-
选择需要的数据库并点击该名称进行下载,本次实验选择rdp_16s_v16.fa.gz和silva_16s_v123.fa.gz数据库;

-
在当前工作区中创建存放数据库的文件夹,这里笔者为了方便管理与查看命名为“3_database”,新建文件夹的命令如下:
mkdir -p 3_database -
将下载后的数据库压缩包移动到该文件夹内,并且进行解压缩
gunzip -c rdp_16s_v16.fa.gz > rdp_16s_v16.fa gunzip -c rdp_16s_v16.fa.gz > rdp_16s_v16.fa
第四步:序列合并和重命名
操作名称
-
序列合并和重命名
-
单端序列:由单端(Single-end(SE))测序产生的测序文件,1个fastq文件。
-
双端序列:由双端(Pair-end(PE))测序产生的测序文件,2个fastq文件分别存放read1和read2的数据。

- 该步骤中的重命名与第二步为序列文件重命名不同的是为序列文件中的每个序列进行重命名。
-
操作原因
- 序列合并:因为测序序列的尾部质量都下降的很严重,如果将双端序列合并,并可以对尾部的低质量序列进行矫正。
- 重命名:对序列文件中每个序列进行重命名,可方便后面构建特征表等操作时方便识别各个序列。
操作方法
-
单个序列文件合并和重命名
序列合并使用vsearch工具的
--fastq_mergepairsfilename选项,该参数是用来将双端序列合并成一个序列文件。完成合并序列的目的还需要搭配--reverse和--fastaout/fastqout选项。选项 参数 –fastq_mergepairs 序列文件1的路径 –reverse 序列文件2的路径 –fastqout 合并序列文件(fastq格式)的输出路径 –fastaout 合并序列文件(fasta格式)的输出路径 而如果要完成序列重命名的操作,还需要使用
--relabel选项来实现重命名。选项 参数 –relabel 给定重新命名的字符串 完成单个序列文件合并和重命名的命令如下:
vsearch --fastq_mergepairs 2_seq/KO1_1.fq.gz \ --reverse 2_seq/KO1_2.fq.gz \ --fastqout 4_merge_and_rename/KO1.merged.fq \ --relabel KO1.笔者为了方便数据的管理,将合并后的序列数据都放在了“4_merge_and_rename”文件夹内,这需要你提前在工作区也新建“4_merge_and_rename”文件夹,
mkdir -p 4_merge_and_rename。如下便是运行完的界面,当看到“Merging reads 100%“便是运行成功,后面是对本次合并的结果进行的统计说明。
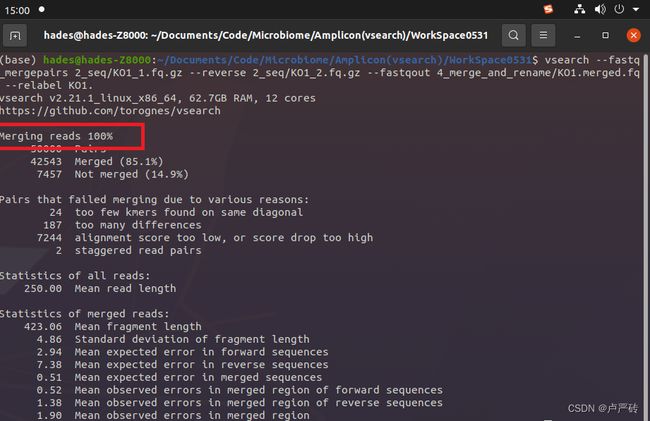
在实际操作中,如果需要完成想要的合并效果可能还需要其他选项进行搭配使用。下面的表格是笔者根据官方手册整理出的选项介绍,供读者选择。
选项 参数 –fastqout_notmerged_fwd 序列文件1中没有合并的序列组成的序列文件(fastq格式)的输出路径 –fastaout_notmerged_fwd 序列文件1中没有合并的序列组成的序列文件(fasta格式)的输出路径 –fastqout_notmerged_rev 序列文件2中没有合并的序列组成的序列文件(fastq格式)的输出路径 –fastaout_notmerged_rev 序列文件2中没有合并的序列组成的序列文件(fasta格式)的输出路径 –eetabbedout 序列合并统计情况的文件的输出路径 –fastq_truncqual 输入正整数。从3’端开始当碱基的质量值Q小于该正整数则剔除该序列,不进行合并 –fastq_minlen 输入正整数(默认是1)。当序列长度小于该正整数时丢弃该序列,不进行合并 –fastq_maxns 输入正整数(默认没有限制)。当序列中模糊碱基(N)的数量超过该正整数时丢弃该序列,不进行合并 –fastq_allowmergestagger 无参数。如果添加该选项,对于交错的序列也会进行合并 –fastq_minovlen 输入正整数(最小为5,默认是10)。作为序列合并重叠区域的最小长度,低于则舍弃序列不进行合并。 –fastq_maxdiffs 输入正整数(默认是10)。当序列合并区域中碱基不匹配的数量大于该正整数时舍弃该序列,不进行合并 –fastq_maxdiffpct 输入小数(默认是100%)。当序列合并区域中碱基不匹配的百分比大于该值时舍弃该序列,不进行合并 –fastq_minmergelen 输入正整数。当合并后的序列长度小于该正整数时丢弃该合并序列 –fastq_maxmergelen 输入正整数。当合并后的序列长度大于该正整数时丢弃该合并序列 –fastq_qminout 输入正整数。确定合并序列输出文件中碱基质量最小的值。 –fastq_qmaxout 输入正整数。确定合并序列输出文件中碱基质量最大的值。 –label_suffix 输入字符串。作为序列名称的前缀。 -
批量序列合并和重命名
我们可以看到在“2_seq”文件夹中,所有文件的命名格式是 “样本名_1.fq.gz” 和 “样本名_2.fq.gz” ,所以我们只需要遍历样本名便可以做到循环获取序列文件,便可以进行批量合并了。
批量的序列合并则还需要采用第二步中批量下载测序数据的思路,不过这一步用到了一个循环执行命令的语句(for循环语句)。
for item in `cat 1_metadata/metadata.csv | csvtk cut -f 1 `; do vsearch --fastq_mergepairs 2_seq/${item}_1.fq.gz \ --reverse 2_seq/${item}_2.fq.gz \ --fastqout 4_merge_and_rename/${item}.merged.fq \ --relabel ${item}. done-
csvtk:cut -f <行数,行数> 参数是挑选特定列的数据,本文是挑选第1列的数据;
-
tail:cat命令可用于查看文件的内容;
-
命令行中for循环语句:
for 变量名 in 列表 do 程序段(command) done -
整个语句的意思是根据cat工具获取样本元数据 ,然后通过管道命令(|)将数据传递给csvtk工具获取第1列数据(样本名),然后将样本名作为列表使用for循环语句进行遍历,每次循环的样本名定义为‘item’,然后套用单个序列文件合并和重命名的命令,替换变量,便可以进行批量序列合并和重命名。
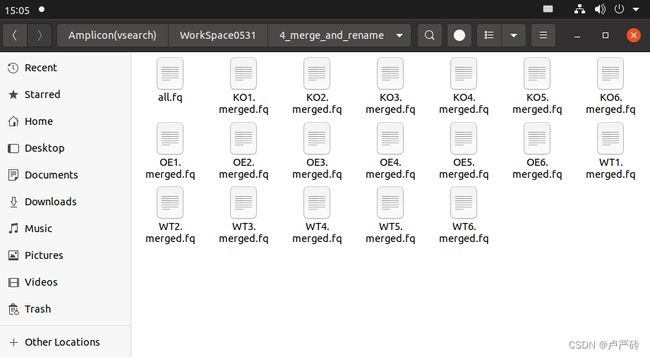
当所有序列合并以后,再将所有的序列文件整合到一个文件中,命令如下:
cat 4_merge_and_rename/*.merged.fq > 4_merge_and_rename/all.fq- 该命令的意思是将所有合并后的序列文件
*.merged.fq内容通过重定向给新的序列文件all.fq
-
数据文件解读

-
第一行:以‘@’开头,是这一条read的名字,在本步骤中已经重命名为“样品名称.序号”的格式;
-
第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
-
第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
-
第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。
第五步:切除引物与质控
操作名称
-
切除序列中的标签和引物并进行质控
对于标签和引物的介绍不在本篇文章中介绍,笔者会在后期单独出一篇文章介绍,需要的读者可以先去看该篇文章。
操作原因
- 切除引物:因为如果需要构建特征表,那么在构建文库过程中加入的引物和标签序列会在序列比对的时候对结果造成很大的影响。所以我们必须在构建特征表之前将引物与标签序列切除掉。
- 质控:对于序列中存在质量值较低的碱基较多时,需移除该序列不进行后续的分析,因为低质量的序列数据也会对后续比对结果造成较大影响。
操作方法
-
序列切除引物与质控使用vsearch工具的
--fastq_filterfilename选项,该参数是用来修剪和筛选给定的序列文件(FASTA或FASTQ格式)然后输出整理后的序列文件(FASTA或FASTQ格式)。完成切除引物和质控的操作还需要搭配--fastq_stripleft、--fastq_stripright和--fastaout/fastqout选项。选项 参数 –fastq_filter 输入字符串。待处理的序列文件路径(fastq格式) –fasta_filter 输入字符串。待处理的序列文件路径(fasta格式) –fastx_filter 输入字符串。待处理的序列文件路径(fasta或fastq格式) –fastq_stripleft 输入正整数。切除上游引物的长度 –fastq_stripright 输入正整数。切除下游引物的长度 –fastq_maxee_rate 输入小数。当序列中所有碱基的错误概率的和除以序列的长度所得的值大于该值时,剔除该序列不进行操作 –fastqout 合并序列文件(fastq格式)的输出路径 –fastaout 合并序列文件(fasta格式)的输出路径 而如果要完成序列重命名的操作,还需要使用
--fastq_maxee_rate选项来实现重命名。选项 参数 –fastq_maxee_rate 输入小数。当序列中所有碱基的错误概率的和除以序列的长度所得的值大于该值时,剔除该序列不进行操作 –fastq_maxee 输入小数。当序列中所有碱基的错误概率的和大于该值时,剔除该序列不进行操作 这里笔者还是建议质控的时候用
--fastq_maxee_rate选项来实现,因为如果只是求和的话,每个序列的长度不同,所以比较起来会有比较大的偏差。完成切除引物与质控的命令如下:
vsearch --fastx_filter 4_merge_and_rename/all.fq \ --fastq_stripleft 29 --fastq_stripright 18 \ --fastq_maxee_rate 0.01 \ --fastaout 5_cut_primers_and_quality_filter/filtered.fa笔者为了方便数据的管理(其实就是强迫症了,如果理解的读者把所有文件放在一个文件夹内也可),将切除引物与质控后的序列数据都放在了“5_cut_primers_and_quality_filter”文件夹内,这需要你提前在工作区也新建“5_cut_primers_and_quality_filter”文件夹,
mkdir -p 5_cut_primers_and_quality_filter。如下便是运行完的界面,当看到“Reading input file 100%“便是运行成功,后面是对本次操作的结果进行的统计说明。

在实际操作中,如果需要完成想要的切除引物与质控效果可能还需要其他选项进行搭配使用。下面的表格是笔者根据官方手册整理出的选项介绍,供读者选择。
选项 参数 –fastqout_discarded 序列文件中丢弃的序列组成的序列文件(fastq格式)的输出路径 –fastaout_discarded 序列文件中丢弃的序列组成的序列文件(fasta格式)的输出路径 –fastq_truncee 输入小数。依次对序列的所有碱基的错误概率进行相加,当结果超过该值的时候截断序列,保留之前序列 –fastq_trunclen 输入正整数。按该数值截断序列,保留较长的序列 –fastq_trunclen_keep 输入正整数。按该数值截断序列,保留较短的序列 –fastq_truncqual 输入正整数。按顺序读取序列碱基的质量值,当质量值小于该值时,截断序列 –fastq_maxlen 输入正整数(默认没有限制)。当序列长度超过该正整数时丢弃该序列 –fastq_maxns 输入正整数(默认没有限制)。当序列中模糊碱基(N)的数量超过该正整数时丢弃该序列 –fastq_minlen 输入正整数(默认没有限制)。当序列长度小于该正整数时丢弃该序列 –eeout 无参数。如果添加该选项,可以在输出序列的名称旁边显示该序列所有碱基的错误概率的和 注:当输入文件时FASTA格式时,一下选项都不可以用
--eeout,
--fastq_maxee,--fastq_maxee_rate,--fastq_out,--fastq_truncee,--fastq_truncqual,--fastqout_discarded。
数据文件解读

-
第一行:以‘>’开头,是这一条read的名字,在本步骤中已经重命名为“样品名称.序号”的格式;
-
第二行:删除标签和引物以后的测序read的序列;
ACGCTCGACAAACAGGATTAGATACCCTG|GTAGTCCACGCCCTAAACGATGTGTGCTGGGCGTCGGGGGGCTTGCCCCTCGGTGCCGGAGCCAACGCGGTAAGCACACCGCCTGGGGAGTACGGCCGCAAGGTTAAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGCGGAGCATGTTGCTTAATTCGACGCAACGCGAAGAACCTTACCAAGGCTTGACATCGCCGGAAAACTCGCAGAGATGCGGGGTCCTTTTGGGCCGGTGACAGGTGGTGCATGGCTGTCGTCAGCTCGTGTCGTGAGATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTCGTTCTATGTTGCCAGCACGCCCTTCGGGGTGGTGGGGACTCATAGGAGACTGCCGGGGTCAACTCGG|AGGAAGGTGGGGAGACGT GTAGTCCACGCCCTAAACGATGTGTGCTGGGCGTCGGGGGGCTTGCCCCTCGGTGCCGGAGCCAACGCGGTAAGCACACCGCCTGGGGAGTACGGCCGCAAGGTTAAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGCGGAGCATGTTGCTTAATTCGACGCAACGCGAAGAACCTTACCAAGGCTTGACATCGCCGGAAAACTCGCAGAGATGCGGGGTCCTTTTGGGCCGGTGACAGGTGGTGCATGGCTGTCGTCAGCTCGTGTCGTGAGATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTCGTTCTATGTTGCCAGCACGCCCTTCGGGGTGGTGGGGACTCATAGGAGACTGCCGGGGTCAACTCGG从前后数据可以看出,处理后的序列数据前后各删除了部分序列,该部分就是标签和前后端引物。
第六步:序列去冗余
操作名称
- 去除序列文件中冗余的序列和低丰度的序列
操作原因
- 去除重复的序列,为后续的聚类或去噪等来寻找特征序列的操作提高计算速度
- 去除低丰度的序列,同样也是为了后续的聚类或去噪等操作提高计算速度
操作方法
-
vsearch对于去冗余功能,提供了四个选项供读者使用。分别是
--derep_fulllength、--derep_id、--derep_prefix和--fastx_uniques。--derep_fulllength和--fastx_uniques的使用场景一致,都是严格识别序列碱基,但是忽略大小写并且碱基T和U不做区分。这里要说明的是--derep_fulllength选项在新版已经不建议使用了,官网建议用--fastx_uniques选项代替。--derep_id的使用场景是除了严格的识别序列,也会识别序列的头文件和名称。--derep_prefix的使用场景是用来所有序列根据名称前缀进行分组来用的。
通过以上的介绍,我们已经可以知道,在序列去冗余操作中我们常用的选项是
--fastx_uniques和--derep_fulllength。但是完成常见的去冗余功能,还需要搭配--minuniquesize、--sizeout、--relabel和--output选项。选项 参数 –derep_fulllength 输入字符串。待处理的序列文件路径(fastq或fasta格式) –fastx_uniques 输入字符串。待处理的序列文件路径(fastq或fasta格式) –derep_id 输入字符串。待处理的序列文件路径(fastq或fasta格式) –derep_prefix 输入字符串。待处理的序列文件路径(fastq或fasta格式) –minuniquesize 输入正整数。在序列去冗余的过程中,如果序列的丰度小于该值时,则移除该序列 –sizeout 无参数。添加该选项后,会在输出文件的每个序列名称后显示该序列的丰度 –relabel 输入字符串。作为序列重命名的字符串 –output 去冗余后序列文件的输出路径,需搭配 --derep_fulllength使用–fastaout 去冗余后序列文件的输出路径,需搭配 --fastx_uniques使用–fastaout 去冗余后序列文件的输出路径,需搭配 --fastx_uniques使用完成序列去冗余的命令如下:
# 方法一--derep_fulllength vsearch --derep_fulllength 5_cut_primers_and_quality_filter/filtered.fa \ --minuniquesize 10 --sizeout --relabel Uni_ \ --output 6_dereplicate/uniques.fa # 方法二:--fastx_uniques vsearch --fastx_uniques 5_cut_primers_and_quality_filter/filtered.fa \ --minuniquesize 10 --sizeout --relabel Uni_ \ --fastaout 6_dereplicate/uniques.fa- 笔者因为强迫症(不装了,摊牌了),将去冗余后的序列数据都放在了“6_dereplicate”文件夹内,这需要你提前在工作区也新建“6_dereplicate”文件夹,
mkdir -p 6_dereplicate。 - 这里的
--relabel选项可以不加,读者是为了和前面的序列区别开,所有重新命名。
如下便是运行完的界面,当看到“Writing FASTA output file 100%“便是运行成功,后面是对本次去冗余的结果进行的统计说明。
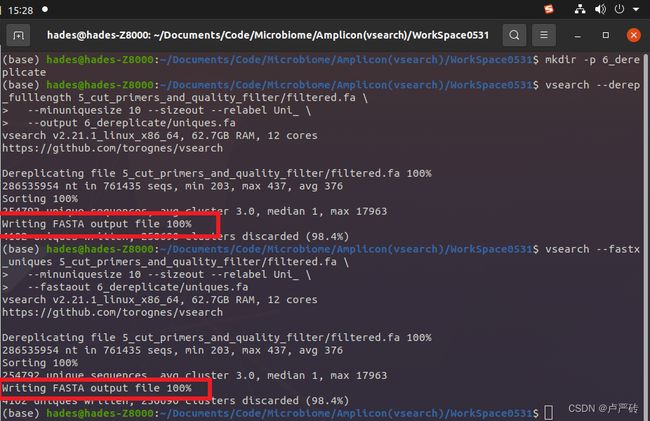
在实际操作中,如果需要完成想要的序列去冗余效果可能还需要其他选项进行搭配使用。下面的表格是笔者根据官方手册整理出的选项介绍,供读者选择。
选项 参数 –fastq_ascii 输入33或64(默认是33)。用于定义输入fastq文件质量分数ASCII码的类型选择,33代表Sanger / Illumina 1.8+ 格式 (从33开始);64代表Solexa,llumina 1.3+ and Illumina 1.5+ 格式(从64开始) –fastq_asciiout 输入33或64(默认是33)。用于定义输出fastq文件质量分数ASCII码的类型选。 –fastq_qmax 输入正整数(默认41)。指定读取fastq文件时接受的最大质量分数。 –fastq_qmaxout 输入正整数(默认41)。指定输出fastq文件时接受的最大质量分数,旧版本最大的质量分数为40。 –fastq_qmin 输入正整数(默认0)。指定读取fastq文件时接受的最小质量分数。 –fastq_qminout 输入正整数(默认0)。指定输出fastq文件时接受的最小质量分数,旧版本最小的质量分数在-5到2之间。 –fastq_qout_max 无参数。如果使用该选项,在输出文件中会输出每个碱基的质量分数。 –maxuniquesize 输入正整数。如果序列的丰度大于该值,则舍弃该序列。 –minuniquesize 输入正整数。如果序列的丰度小于该值,则舍弃该序列。 –relabel 输入字符串。作为序列重命名的字符串 –relabel_keep 无参数。如果使用该选项,当重命名时旧的标题将保留在新名称后面。 –relabel_md5 无参数。添加该选项后,会使用MD5算法对序列计算,并把计算结果作为序列的标题。 –relabel_self 无参数。添加该选项后,会使用序列本身作为序列标题。 –relabel_sha1 无参数。添加该选项后,会使用SHA1算法对序列计算,并把计算结果作为序列的标题。 –sizein 无参数。添加该选项后,将读取输入文件中的每个序列的丰度注释(格式为‘[>;]size= integer[;]‘) –sizeout 无参数。添加该选项后,会在输出文件的每个序列名称后显示该序列的丰度 –strand 输入“plus”或“both”。当参数为“plus”时,比较两个序列时,只检查主要的内个序列;当参数为“both”时,则同时检查两个序列。 –topn 输入正整数。输出丰度最高的前几位序列。 –xsize 无参数。添加该选项后,如果输入文件中有丰度注释,则不输出该丰度注释。
数据文件解读
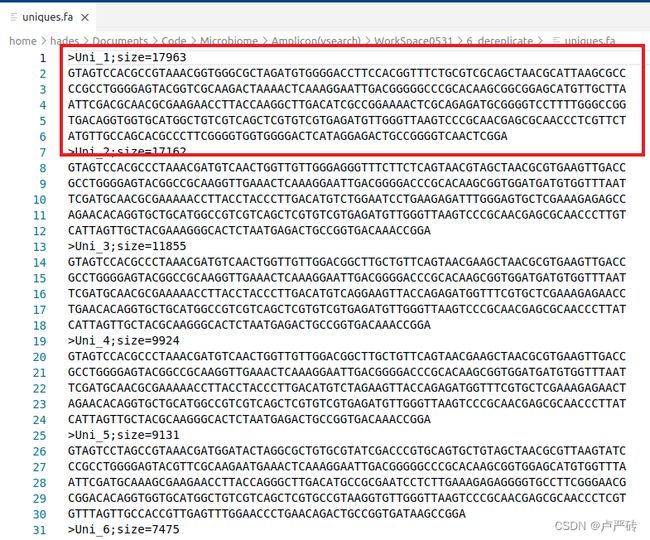
- 第一行:以‘>’开头,是这一条read的标题和备注信息,在本步骤中已经重命名为“Uzi_序号”的格式(该序号是进过丰度由大到小排序后的序列),分号(;)后面说明的是该序列的丰度,由“size=丰度”表示;
- 第二行:序列内容。
第七步:特征挑选
操作名称
- 挑选特征序列(代表性序列)
- 聚类
- OTU通常按97%聚类后挑选最高丰度或中心的代表性序列;
- 去噪
- ASV是基于序列进行去噪(排除或校正错误序列,并挑选丰度较高的可信序列)作为代表性序列
- 聚类
操作原因
-
高通量测序得到的16S序列有成千上万条,如果对每条序列都进行物种注释的话,工作量大、耗时长,而且16S扩增、测序等过程中出现的错误会降低结果的准确性。在16S分析中引入OTU,首先对相似性序列进行聚类,分成数量较少的分类单元,基于分类单元进行物种注释。这不仅简化工作量,提高分析效率,而且OTU在聚类过程中会去除一些测序错误的序列,提高分析的准确性。
-
因为去冗余后的序列仍然远多于物种数量,并且扩增的物种可能存在rDNA的多拷贝且存在变异而得到来自同一物种的多条序列扩增结果。
多拷贝基因:多拷贝基因(duplicate gene)是指进化过程中,高等生物的基因组会发生大量重复。这些重复DNA序列有的继续发生进化歧异,成为与原来序列不同的新基因;有的以结构和功能仍基本相同的形式保留下来成为多拷贝基因。
代表性序列(representative sequences)即为确定的最终版的OTU,类似于参考基因组/cDNA将为索引的字典。然后将所有数据mapping于OTU上来确定各物种的丰度。
操作方法
-
聚类
-
vsearch关于聚类的方法提供了三个选项,分别是
--cluster_fast、--cluster_size和--cluster_smallmem。--cluster_fast选项适用于按序列长度排序后进行聚类的情况;--cluster_size选项适用于按序列丰度排序后进行聚类的情况;--cluster_smallmem选项适用于省内存方式聚类,默认按长度进行聚类,除非指定—usersort选项,否则提前不按照丰度排序。
通过以上介绍,已经可以知道vsearch常见的聚类选项,但是完成常见的聚类操作还是需要搭配
--id、--iddef、--centroids和--relabel选项。选项 参数 –cluster_fast 输入字符串。待处理的序列文件路径,按序列长度排序聚类 –cluster_size 输入字符串。待处理的序列文件路径,按序列丰度排序聚类 –cluster_smallmem 输入字符串。待处理的序列文件路径,省内存方式聚类,不按丰度排序,默认按长度,除排指定 --usersort—usersort 无参数。添加该选项后,允许任何序列输入排序,不只是按照长度排序。 –id 输入0-1之间的小数。作为相似度阈值 –iddef 输入0、1、2、3、4。每个数字代表一个相似度计算模式,默认模式为2 –centroids 输出的代表序列文件位置。 –relabel 输入字符串。作为序列重命名的字符串 –sizeout 无参数。添加该选项后,会在输出文件的每个序列名称后显示该序列的丰度 相似度计算模式介绍如下:
模式代号 模式详情 0 CD-HIT工具定义:匹配长度/最短序列长度 1 作者定义:匹配长度/代表序列长度 2 作者定义:匹配长度/(代表序列长度-端子间隙) 3 海洋生物实验室定义:1.0-[(未匹配长度+间隙开口)/(最短序列长度)] 4 BLAST工具定义:匹配长度/最短序列长度 完成聚类的命令如下:
vsearch --cluster_fast 6_dereplicate/uniques.fa \ --id 0.97 \ --centroids 7_cluster/otu.fa \ --relabel OTU_ \ --sizeout如下便是运行完的界面,当看到“Writing clusters 100%“便是运行成功,后面是对本次操作的结果进行的统计说明。其中可以看到共挑选了1073个特征序列,其中有499个Singletons,占总特征序列的12.2%。
Singletons:单次出现序列。根据Singletons的占比,可以为后续多样新分析提供参考。

- 这里笔者将挑选好的特征序列数据都放在了“7_cluster”文件夹内,这需要你提前在工作区也新建“7_cluster”文件夹,
mkdir -p 7_cluster。 - 这里的
--relabel选项可以不加,读者是为了和前面的序列区别开,所有重新命名。
-
-
去噪
-
vsearch的去噪功能通过
--cluster_unoise选项完成,并且完成常见的去噪功能还需要搭配--unoise_alpha、--minsize、--centroids和--relabel选项。选项 参数 –cluster_unoise 输入字符串。待去噪的序列文件路径 –unoise_alpha 输入小数(默认参数为2.0)。 --cluster_unoise命令的子参数,因为算法比较复杂,笔者查看论文初步了解到,如果改值越大,排除或校正错误序列的标准就越严格–minsize 输入正整数(默认参数为8)。改选项用于指定序列去噪时的最小丰度 –centroids 输出代表序列的文件位置。 –relabel 输入字符串。作为序列重命名的字符串 –sizeout 无参数。添加该选项后,会在输出文件的每个序列名称后显示该序列的丰度 完成去噪功能的命令如下:
vsearch --cluster_unoise 6_dereplicate/uniques.fa \ --minsize 10 \ --centroids 7_denoise/asv.fa \ --relabel ASV_ \ --sizeout如下便是运行完的界面,当看到“Writing clusters 100%“便是运行成功,后面是对本次操作的结果进行的统计说明。其中可以看到共挑选了3145个特征序列,其中有2928个Singletons,占总特征序列的71.4%。
- 这里笔者将挑选好的特征序列数据都放在了“7_denoise”文件夹内,这需要你提前在工作区也新建“7_denoise”文件夹,
mkdir -p 7_denoise。 - 这里的
--relabel选项可以不加,读者是为了和前面的序列区别开,所有重新命名。
在实际操作中,如果需要完成想要的特征挑选的效果可能还需要其他选项进行搭配使用。下面的表格是笔者根据官方手册整理出的选项介绍,供读者选择。
选项 参数 –biomout 输入字符串。输出biom格式1.0的OTU表的路径,JSON文件,详见http://biom-format.org/documentation/format_versions/biom-1.0.html –clusterout_id 无参数。当使 --centroids、--consout和--profile时,添加簇的标志符信息–clusterout_sort 无参数。当使 --centroids、--consout,--msaout、--profile和--uc时,结果按丰度降序排列–clusters 输入字符串。以字符串为前缀,输出结果为每条序列一个fasta文件 –consout 输入字符串。输出每个cluster比对的一致序列 –msaout 输入字符串。输出多序列比对的fasta文件 –mothur_shared_out 输入字符串。输出mother格式的OTU表 –otutabout 输入字符串。输出经典表格格式的 OTU表 –profile 输入字符串。输出多序列比对频率谱文件 –qmask 输入none|dust|soft(默认参数为dust)。选择屏蔽序列的方法。当使用soft方式时聚类将会区分大小写 –qsegout 输入字符串。将所有的查询序列的对齐部分输出为fasta文件 –sizeorder 无参数。添加该参数时,如果扩增子有多个可能中心时,考虑丰度优先 –tsegout 输入字符串。将所有的目标序列的对齐部分输出为fasta文件 –uc 输入字符串。输出uclust结果格式的文件 - 这里笔者将挑选好的特征序列数据都放在了“7_denoise”文件夹内,这需要你提前在工作区也新建“7_denoise”文件夹,
-
数据文件解读
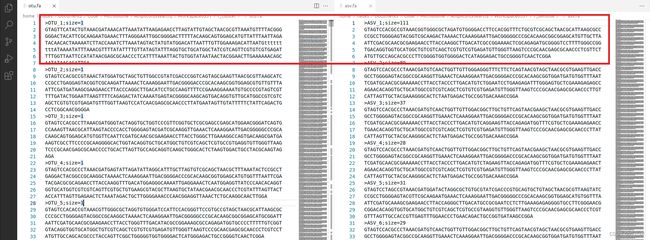
- 第一行:以‘>’开头,是这一条read的标题和备注信息,在本步骤中已经重命名为“OTU_序号”或“ASV_序号”的格式。
- 第二行:序列内容。
第八步:嵌合体检测
操作名称
-
嵌合体检测
- 序列自身比对去嵌合体
- 基于参考数据库去嵌合体
-
嵌合体(Chimeras) 是PCR阶段不正常的扩增过程所产生的序列。
嵌合体由两条及以上的模板链组成,是PCR延伸阶段的不完全延伸造成的。 -
通常有1%的几率会出现嵌合体序列,而在16S/18S/ITS扩增子测序的分析中,由于序列间相似度较高,所以概率会更高。
-
在去除嵌合体的同时我们会造成一定的数据损失,是否需要去除嵌合体是一个有争议的问题,如果你不想去除嵌合体可以选择跳过这一步直接进行后续的分析。
-
嵌合体序列由来自两条或者多条模板链的序列组成,如图所示,在扩增序列X的过程中,在序列延伸阶段,只产生了部分X序列延伸阶段就结束了,在下一轮的PCR反应中,这部分序列作为序列Y的引物接着延伸,扩增就会形成X和Y的嵌合体序列

操作原因
- 通常在PCR过程中,大概有1%的几率会出现嵌合体序列,而以在16S/18S/ITS 扩增子测序的分析中,系统相似度极高,嵌合体可达1%-20%,需要去除嵌合体序列。嵌合体的比例与PCR循环数相关,循环数越高,嵌合体比例越高。事实上嵌合体在正常生物体中是不存在的,所以在16S扩增子测序的分析中,需要去除嵌合体序列。
- 如果存在大量嵌合体序列,那么将会对后续生成特征表时,让研究人员进入不必要的误区,所以需要检测嵌合体并去除。
操作方法
-
序列自身比对去嵌合体
-
vsearch关于序列自身比对去嵌合体的方法提供了三个选项,分别是
--uchime_denovo、--uchime2_denovo和--uchime3_denovo。vsearch在进行序列自身比对去嵌合体时,输入文件必须时带有丰度注释的序列文件(在每个序列名称后有“[;]size= integer[;] ”格式的丰度注释)。--uchime_denovo选项是按照序列丰度排列,根据自身去嵌合,无需参考数据库(不支持多线程);--uchime2_denovo选项也是按照序列丰度排列,根据UCHIME2去嵌合(不支持多线程);--uchime3_denovo选项与选项--uchime2_denovo类似,唯一不同的就是默认的最小丰度偏差(--abskew)是16,而--uchime2_denovo是2。
当然完成常见的序列自身比对去嵌合体的操作还是需要搭配
--nonchimeras选项。选项 参数 –uchime_denovo 输入字符串。待处理的序列文件路径 –uchime2_denovo 输入字符串。待处理的序列文件路径 –uchime3_denovo 输入字符串。待处理的序列文件路径 –abskew 输入小数。当完成序列自身比对去嵌合体时,丰度偏差(abundance skew)是三路比较方法中用于区分嵌合体和亲本的标准,该值的意义是需要亲本丰度是嵌合体丰度的多少倍。uchime3中默认值为16,其他情况中默认值为2 –nonchimeras 输入字符串。无嵌合体结果文件的输出路径 完成序列自身比对去嵌合体的命令如下:
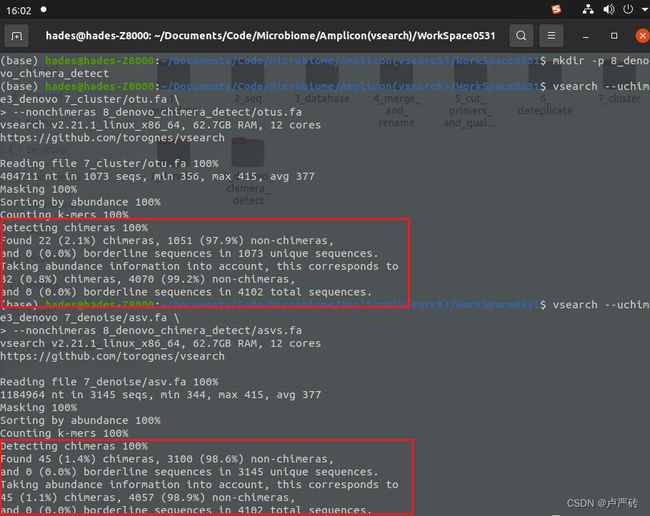
# otu去嵌合体 vsearch --uchime3_denovo 7_cluster/otu.fa \ --nonchimeras 8_denovo_chimera_detect/otus.fa #asv去嵌合体 vsearch --uchime3_denovo 7_denoise/asv.fa \ --nonchimeras 8_denovo_chimera_detect/asvs.fa这里笔者将去嵌合体后的序列数据都放在了“8_denovo_chimera_detect”文件夹内,这需要你提前在工作区也新建“8_denovo_chimera_detect”文件夹,
mkdir -p 8_denovo_chimera_detect。如下便是运行完的界面,当看到“ 100%“便是运行成功,后面是对本次操作的结果进行的统计说明。其中可以看到嵌合体数量等信息。
-
-
基于参考数据库去嵌合体
-
vsearch想要实现基于参考数据库去嵌合体的操作需要使用
--uchime_ref选项。当然如果要完成操作还是需要搭配--db和--nonchimeras选项。选项 参数 –uchime_ref 输入字符串。待处理的序列文件路径 –db 输入字符串。当使用 --uchime_ref选项时,需使用该选项指定数据库fasta文件的位置–nonchimeras 输入字符串。无嵌合体结果文件的输出路径 本次基于参考的数据库选择silva数据库,因为数据库的选择越大,假阴性率就越低;当然你也可以选择rdp数据库进行实验。完成基于参考数据库去嵌合体的命令如下,:
# otu去嵌合体 vsearch --uchime_ref 7_cluster/otu.fa \ --db 3_database/silva_16s_v123.fa \ --nonchimeras 8_reference_chimera_detect/otus.fa # asv去嵌合体 vsearch --uchime_ref 7_denoise/asv.fa \ --db 3_database/silva_16s_v123.fa \ --nonchimeras 8_reference_chimera_detect/asvs.fa这里笔者将去嵌合体后的序列数据都放在了“8_reference_chimera_detect”文件夹内,这需要你提前在工作区也新建“8_reference_chimera_detect”文件夹,
mkdir -p 8_reference_chimera_detect。如下便是运行完的界面,当看到“Writing clusters 100%“便是运行成功,后面是对本次操作的结果进行的统计说明。其中可以看到嵌合体数量等信息。

在实际操作中,如果需要完成想要的去嵌合体效果可能还需要其他选项进行搭配使用。下面的表格是笔者根据官方手册整理出的选项介绍,供读者选择。
选项 参数 –self 无参数。当使用 --uchime_ref选项时,忽略原始和数据库中的同名序列–selfid 无参数。当使用 --uchime_ref选项时,忽略原始和数据库中完全相同的序列–chimeras 输入字符串。输出嵌合体序列的文件路径 –borderline 输入字符串。输出无法确定的嵌合体的文件路径。它们像嵌合体,但不足以区分其和亲本 –uchimeout 输入字符串。输出嵌合体比对详细结果的文件路径 –uchimeout5 输入字符串。输出嵌合体比对详细结果(usearch5版本格式)的文件路径 –uchimealns 输入字符串。输出嵌合体三路比较结果的文件路径 –alignwidth 输入正整数。当使用 --uchimealns选项时,设置该参数为三路比对的宽度,默认为80,0为无限制–fasta_score 无参数。当使用该参数时,会在输出的嵌合体序列结果中包含嵌合体打分,打分相关的五个选项为( --dn,--mindiffs,--mindiv,--minh,--xn)–dn 输入正实数(默认值为1.4)。该选项对应于嵌合体评分函数的参数n –mindiffs 输入正整数(默认值为3)。 –mindiv 输入小数(默认值为0.8)。该参数表示每部分的最小不同, --uchime2_denovo和--uchime3_denovo中此参数无效–minh 输入小数(默认值为0.28)。该选项表示与亲本最小分歧, --uchime2_denovo和--uchime3_denovo中此参数无效–xn 输入正实数(默认值为8.0)。该选项对应于嵌合体评分函数的参数beat
-
第九步:生成特征表
操作名称
-
生成特征表
特征表,也可以叫做OTU表,是每个特征序列在每样品中的丰度值,本质上每种高通量测序结果,都会有一个类似的表,如RNA-Seq是基因表达与样品的表
操作原因
- 微生物多样性分析中最基础、最重要的文件为OTU table,几乎所有的后续分析,如alpha多样性分析,beta多样性分析,差异分析等等都是基于OTU table展开的。
操作方法
-
vsearch工具提供了具有全局搜索功能的
--usearch_global选项,来完成OTU代表序列与微生物数据库比对的任务。但是想要完成比对后输出特征表还需要搭配--db、--id和--otutabout选项。选项 参数 –usearch_global 输入字符串。待处理的序列文件路径 –db 输入字符串。当使用 --usearch_global参数时,设置该参数说明指定数据库或参考序列文件(fasta格式)的文件路径。–id 输入0-1之间的小数。作为相似度阈值 –iddef 输入0、1、2、3、4。每个数字代表一个相似度计算模式,默认模式为2 –threads 输入1-1024之间的正整数。输入的参数作为计算使用的线程数,但是参数必须小于或等于你的CPU核心数。默认情况下将所有的核心都用到,每个核心创建一个线程。 –otutabout 输入字符串。输出经典表格格式的 OTU表的文件路径 使用vsearch工具生成特征表的命令如下(本例以基于参数去嵌合的OTU序列作为参考序列文件,读者可修改
--db选项参数切换其他的特征序列参考文件):vsearch --usearch_global 5_cut_primers_and_quality_filter/filtered.fa \ --db 8_reference_chimera_detect/asvs.fa \ --id 0.97 \ --threads 4 \ --otutabout 9_otu_table/otutab.txt如下便是运行完的界面,当看到“Writing FASTA OTU table 100%“便是运行成功,其他是对本次操作的结果进行的统计说明。
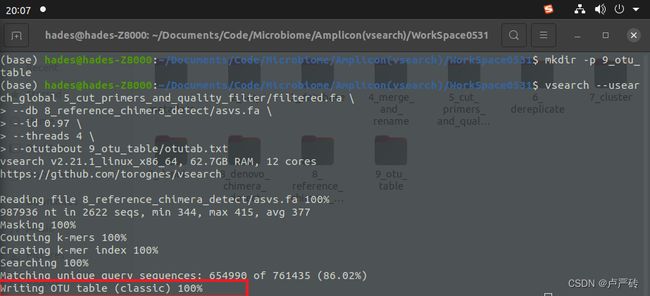
这里笔者将特征表都放在了“9_otu_table”文件夹内,这需要你提前在工作区也新建“9_otu_table”文件夹,
mkdir -p 9_otu_table。
数据文件解读
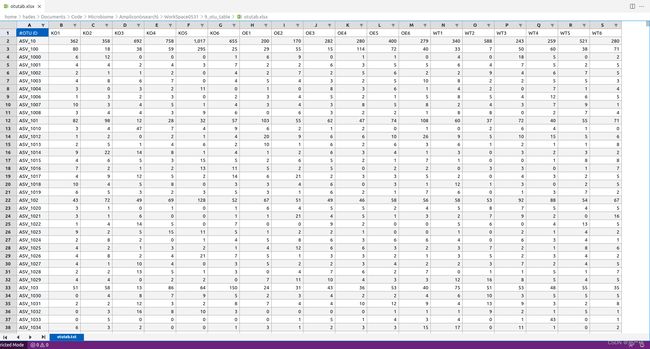
- 横向表头为样本名
- 纵向表头为特征序列
- 表的内容是每个特征序列在该样本中丰度值。
第十步:物种注释
操作名称
- 将OTU代表序列与相应的微生物数据库比对(Silva、RDP、Greengene等),得到每个样本所含的物种信息。
操作原因
- 对于扩增子分析,最重要的就是物种信息。所以基于去嵌合体的代表序列,使用具有物种注释信息的参考序列或者数据库进行物种注释很有必要。
操作方法
-
vsearch工具提供了
--sintax来实现物种的类别注释,但是要完成物种注释还需要搭配--db、--sintax_cutoff、--tabbedout和--randseed选项。选项 参数 –sintax 输入字符串。待处理的序列文件路径 –db 输入字符串。当使用 --sintax参数时,设置该参数说明指定具有物种注释信息的数据库或参考序列文件(fasta格式)的文件路径。–sintax_cutoff 输入小数。为输出的注释文件中预测分类列指定bootstrap,当预测bootstrap小于该参数,那么删除该注释信息 –tabbedout 输入字符串。输出的物种注释文件的文件路径 –randseed 输入正整数(默认是0)。该参数作为算法中使用的随机数生成器的种子,默认0是作为一个伪随机种子 使用vsearch工具完成物种注释的命令如下:
vsearch --sintax 8_reference_chimera_detect/asvs.fa \ --db 3_database/silva_16s_v123.fa \ --sintax_cutoff 0.1 \ --tabbedout 10_taxonomic_classification/asvs.sintax这里笔者将物种注释文件放在了“10_taxonomic_classification”文件夹内,这需要你提前在工作区也新建“10_taxonomic_classification”文件夹,
mkdir -p 10_taxonomic_classification。如下便是运行完的界面,当看到“Classifying sequences 100%“便是运行成功,其他是对本次操作的结果进行的统计说明。

数据文件解读
- 物种注释文件为“.sintax”文件格式,该文件由多条数据组成。
- 每条数据首先是特征序列名称;然后后面的一串数据直到“+”结束,这段数据表示特征序列的物种类别和置信度(括号内的就是置信度);“+”后面的一串数据是没有置信度的物种类别。
- 可以通过该物种注释文件,生成标准两列注释文件和八列注释文件,在这里笔者不详细展开了,笔者会在后面的分析文章中详细说明。
以上就是在扩增子分析流程中,使用vsearch工具进行前期数据处理的方法。笔者也是因为上研究生的科研方向涉及到这些知识才学习的,也是刚接触,生物相关的知识储备也不是很充足,所以在阅读中遇到问题或者错误,可以及时与笔者沟通,笔者会尽快修改。
相关资源
数据处理过程文件
参考文章
https://github.com/YongxinLiu/EasyAmplicon
https://bioinf.shenwei.me/csvtk/