2020-12-14 论文阅读
Deep learning with multimodal representation for pancancer prognosis prediction
2019年发表于Bioinformatics杂志,论文传送门
代码传送门
摘要
动机 估计肿瘤患者未来的病程对于医生来说是非常有价值的;目前临床上肿瘤患者拥有大量的多模态数据,然而现有的方法无法有效地利用这些多模态数据。为了解决这一问题,我们建立了一个多模神经网络模型,用来预测肿瘤患者的生存,研究使用了20种肿瘤的临床数据、mRNA表达数据、miRNA表达数据以及数字病理图像数据。研究开发了一种无监督的编码器,将每个病人的这四种模态的数据压缩为一个向量,我们使用一种可复原的多模态dropout方法处理缺失数据。针对每种数据类型,我们定制了不同的编码方法,对于临床数据和基因组学数据,我们使用了highway networks提取特征,对于数字病理图像,我们使用卷积神经网络提取特征。
结果 我们使用泛癌数据训练了这些特征编码器,预测单一肿瘤以及泛癌的总体生存,达到了0.78的c-index。研究结果表明,建立泛癌预后预测模型,用来预测单一肿瘤的预后是可行的。此外,我们的模型能够处理多模态数据,有效分析数字病理图像数据,将患者的多模态数据转变为一种无监督的,富含信息的特征表示。我们因此提出一种强大的自动化工具,用于准确预测患者预后,这使得我们向着肿瘤个性化治疗迈出了关键一步。
前言
估计肿瘤的进展或者进行预后预测能够有效辅助医生对肿瘤患者的治疗做出决策。为了评估这些患者的预后,医生可以利用多种不同类型的数据,比如临床数据、基因组学数据、病理组织图像以及放射学影像,具体参考的数据依赖于患者所患的肿瘤。但是,这些数据本身属于高维数据,医生凭借自己的能力人工从这些多模态数据中获取到能够解释和预测患者预后的信息,并从中得出治疗决策是非常困难的。其次,肿瘤患者间的异质性决定了肿瘤个体特征对于改善治疗过程是至关重要的。以前的研究表明可以利用机器学习方法对诸如基因表达等分子数据进行挖掘,以对治疗的结局和预后进行预测。类似的研究还发现,使用计算机视觉方法对病理组织图像进行定量分析,可以提取到病理专家所观察不到的额外信息。因此,使用自动化的机器学习系统对高维数据进行分析,挖掘出特征,对于估计疾病的侵袭性和患者的结局有着重要的作用。另外,肿瘤患者间的异质性还提示不同类型的肿瘤之间可能会存在共性。因此,从泛癌数据中提取出这些共性,这将改善模型。TCGA这样的数据库收录了来自多个机构的数据,这些数据包括临床数据、基因组学数据和图像数据,这对于泛癌模型的构建是非常重要的。
然而,由于数据的异质性和高维性质,自动化的预后预测仍然面临着挑战。比如,在TCGA数据库中,一个患者就有成千上万的基因(microRNA或者mRNA)和超高分辨率的数字病理图像(WSI)。虽然基因成千上万,但是根据以往研究,仅有一部分基因或者图像特征和预后是相关的。因此,为了能够成功开发出一种用于预测预后的多模态模型,我们需要一种能够有效处理临床数据、基因数据和图像数据的方法,这些数据本质上是多模态数据。因此,我们开发了一种泛癌深度学习模型结构用来解决这个具有挑战性的问题,我们借鉴了无监督学习和特征表示技术,我们的模型能够充分利用大规模的基因组学和影像数据。本研究的主要目的是使用大量现有的TCGA数据开发出一种稳健的肿瘤特征表示,人们可以通过各种不同的评价指标对这些特征表示进行聚类和比较等。使用无监督的特征表示技术,我们使用包含临床数据、基因组学数据和数字病理图像数据在内的多模态数据开发了一种泛癌生存模型。
背景
我们可以将预后预测表述为删失数据生存分析问题,预测某一事件(如死亡)在给定时间内是否发生以及何时发生。由于生存数据具有独特性,因此一般使用诸如Cox回归分析中的风险函数对其进行表示。
近年来,许多不同的研究方法都试图利用基因组学数据对肿瘤的预后进行预测。比如论文使用一种增强的Cox回归模型对TCGA数据库中的恶性胶质瘤基因表达数据进行生存分析,C-index为0.725。基因数据,尤其是MicroRNA数据已经在多项研究中被认为和预后有关,有研究使用随机森林方法对肾癌MicroRNA数据进行分析,在预后预测上的C-index为0.77。然而,尽管单独使用分子数据进行机器学习建模就能取得不错的效果,但是仍然还有一定的提升空间;毕竟肿瘤微环境非常复杂,很难单独通过分子谱数据对其进行全面的描述。
近年来,许多研究已经证明数字病理图像可以被用来提高预后预测的表现和通用性。由于数字病理图像是由细胞及其周围环境构成的高分辨率图像,其中可能只有一小部分区域与预后有关,因此许多研究都采用一些组合的方法,他们往往由病理学家标注出感兴趣区域(ROIs)。比如论文使用病理专家标注出的ROIs和人为设计的特征进行预后预测,达到了和单独使用基因组学数据建模的表现。最近,深度学习大大提高了预测能力。比如有研究使用卷积神经网络对病理专家标注出的ROIs进行分析,在两个肺癌数据集上进行预后预测,结果超过了利用分子数据的方法。论文和论文发现组织病理图像中存在重要的预后信息,这些信息能够和分子数据相互补充。但是多模态预后预测模型仍然很少有人进行过研究。据我们所知,只有一篇论文将基因组学数据和影像数据进行了结合预测肺癌预后,单独基因组学预测的C-index为0.660,基于数字病理图像ROIs预测的C-index为0.613,将二者结合预测的C-index为0.691。
另外,以上基于数字病理图像的方法需要医生手工标注ROIs,标注过程非常繁琐。自动化的多模预后预测模型中,最难的部分要数找出预后相关的ROIs。在使用数字病理图像进行分类的研究中,有研究提出一种"decision-fusion"模型随机采样图像块然后组合滤波获得分类结果。另外,还有研究使用注意力机制找出重要的图像块。然而,在预后预测方面,基于标注的数字病理图像的方法或者系统取得的成功是有限的。有研究曾使用无监督方法对数字病理图像进行分析,它使用的是K-means聚类的方法,适应性地进行图像块的采样,在肺癌数据上达到了0.708的C-index,这一结果媲美单纯使用基因组学数据的方法。
以前的研究主要集中在单一类型的肿瘤,没有研究不同类型肿瘤之间的共性和联系。尽管之前的研究有使用基因组学数据的,也有使用数字病理图像数据的,但是将二者进行结合的多模态模型少之又少。通过探索多模态数据,自动对数字病理图像进行评分,从中提取出有用的信息,我们或许能够提升现有方法的性能。
近年来,卷积神经网络显著提高了机器学习任务的表现,这些任务包括基因组学数据缺失值估计和基于数字病理图像的预后因子预测。卷积神经网络取得成功的一个重要原因是它能够处理高维的,非结构化的数据,尤其是图像数据。比如卷积神经网络能够通过学习一系列层级特征将不同类别的图像进行很好的分类。即使对于一些图像来说,其输入的大部分像素都被舍弃掉了,卷积神经网络模型依旧能够很好的应对。
预后预测任务要比传统的深度学习任务更加的复杂;我们需要对临床数据、基因数据和数字病理图像数据进行整合预测生存时间,而不仅仅是对相对较小的图像进行分类。此外,由于我们使用的是泛癌数据,所以会有数据缺失情况的出现,比如一些患者可能会缺失影像数据,临床数据或者基因数据,这使得我们很难直接使用标准的卷积神经网络进行分析。无监督学习有着不错的潜力,通过以无监督的方式学习图像特征和基因组学特征,这一过程可以解决标签不足的问题。类似的,表示学习技术能够使得我们探索多模态数据之间的关联性和共性。在预后预测中,至关重要的步骤是将类似的患者映射到相同的抽样特征空间。于是我们提出使用无监督学习和特征表示技术来解决多模态数据建模过程中遇到的一些问题。
材料和方法
数据集和工具
我们的数据主要来自于PanCanAtlas TCGA中(论文)经过预处理和批次校正的数据。这个数据集的数据包括1881个microRNA,60383个基因表达数据,一系列临床信息,我们所使用的临床信息主要为种族,年龄,性别和组织学分级,还包括11000多个患者的数字病理图像。
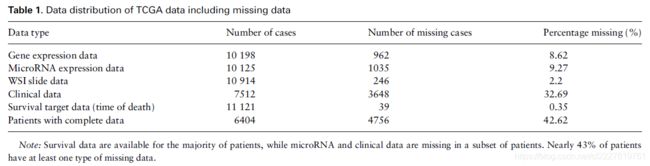
数据集中许多患者都存在数据缺失,所以就有必要保证我们的模型有能力处理这些缺失数据。每个患者都有记录死亡时间,以及删失状态和时间。这20种肿瘤有着不同的生存曲线,如下图:
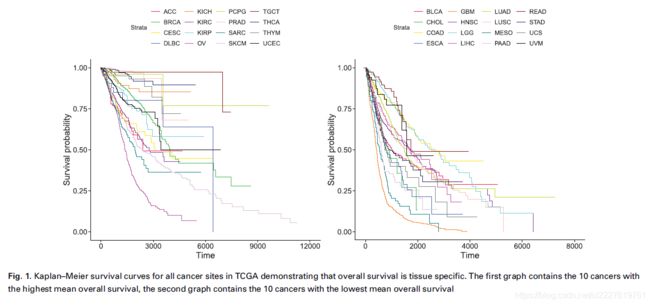
左图中的10种肿瘤平均总体生存时间较长,右图10种肿瘤平均总体生存时间较短。研究使用了openslide模块进行数字病理图像的读取和解析,使用PyTorch深度学习框架进行模型的构建。使用了NVIDIA GTX 1070 GPU进行模型训练。
TCGA数据集的11160名患者被按照85/15的比例划分为训练集和测试集,划分的时候按照每种肿瘤的比例分布进行分层,以确保训练集和测试集中每种肿瘤的比例分布相同。
深度无监督特征学习
为了训练泛癌模型用于预后预测,我们首先尝试将每个患者的多模态数据压缩为一个向量。以前的研究发现不同类型的数据之间(基因表达数据、临床数据、microRNA数据和图像数据)存在着显著的关联,而且以无监督的方式学习这些关联能够显著改善预后的预测过程。因此,本研究采用了一种表示学习框架来指导我们的方法。对于不同类型的数据,尽管自编码器能够收敛,但是它依赖于重构误差,这对于多种来源的异质性数据不是一个很好的选择。相反,我们受论文的启发,在这篇论文中,它将同一物体的两个不同角度的图像输入到孪生网络创建特征表示。对于来自同一物体的不同角度的图像,它们的特征表示之间的余弦相似性被最大化,而对于来自不同物体的图像,它们的特征表示之间的余弦相似性被最小化。为了保持运算稳定性,采用了hinge-loss。这使得来自单个患者的不同数据的特征表示拥有相似性,避免所有患者的特征表示都一样。
在本研究中,我们在上述那篇论文的基础上对公式做了一些修改。我们使用的不是孪生网络,相反,考虑到多模态数据,对于每一种类型的数据,我们都使用了深度学习的方法,但是每一种类型数据的深度学习方法有所不同。

根据经验,我们将特征空间的维度定义为512维。由于我们有两种以上的多模态数据,因此我们将相似性损失进行加和。研究将相似性损失定义 l s i m ( θ ) l_{sim}(\theta) lsim(θ)如下:

其中 x i x_i xi是模态 i i i的数据, h ^ θ , i \hat{h}_{\theta,i} h^θ,i表示模态 i i i所使用的预测模型。注意 M M M控制的是不同特征表示之间的“紧密程度”。 M M M如果较大,那么将允许不同模态数据的特征向量之间存在较大的不同;如果 M M M较小,则表示不同模态数据的特征向量非常相似,这通常是我们希望的一种情况,但是它也会导致模态坍塌(mode collapse)。基于我们的数据,将 M M M所能设置的不发生坍塌的最小值为0.1。我们要对一个batch内的每一对患者计算损失。因此,该无监督模型必须要从基因组学和影像组学数据中识别出能够区分不同患者的重要特征。它也必须要学习一种模态中的特征是如何与另一种模态中的特征相对应的,所以它能为两者产生相似的编码。因此这种方式学习到的特征对于缺失数据是有弹性的。
预后预测
模型除了要学习特征表示之外,还必须能够准确预测预后。因为这是一个生存数据问题,我们的目标是最大化C-index。以前的研究定义了Cox损失函数,它通过优化Cox偏似然函数来进行。因此我们在最后添加了一层结构,将512维的特征向量转变为生存时间。我们使用标准的Cox损失来优化模型,Cox损失定义如下:

其中 T i T_i Ti表示生存时间, E i E_i Ei表示删失状态, x i x_i xi表示每个患者的数据, h θ ^ \hat{h_\theta} hθ^表示输入数据输出生存时间的神经网络模型。结合之前的无监督模型,总的损失为:
![]()
模型结构
针对每一种类型的数据,我们专门使用一种网络结构。对于临床数据,我们使用全连接层,sigmoid激活函数以及dropout。对于基因数据和microRNA数据,我们使用highway networks网络结构。由于数字病理图像的复杂性,我们使用卷积神经网络结构对图像数据进行编码。下面将详细介绍每种网络结构。
每个患者的基因组学和microRNA数据是以密集的一维向量表示的,传统做法通常不采用神经网络对其进行分析,通常采用的是支持向量机或者随机森林方法。但是,为了在优化相似损失和Cox损失的过程中确保模型可微分,我们必须要使用神经网络进行特征提取。近期的研究也表明了深度学习技术的有效性,因此我们使用一种highway network结构训练10层神经网络,这种结构能够避免梯度消失的发生。
为了表示和编码数字病理图像,我们需要开发一种能够有效对数字病理图像进行有效“总结”的机器学习方法。但是高分辨率的数字病理图像使得从整体上对它们学习变得很困难。因此我们必须使用随机采样和滤波的方式。本研究使用了一种相对简单的采样方式来获取ROIs,我们从最高分辨率下采样200个 224 × 224 224\times224 224×224 pixel的图像块,然后计算每个图像块的“color balance”,即使用均方误差来表示每个图像块的RGB三通道颜色均值与整个数字病理图像的RGB三通道的颜色均值的差距。接下来,我们再从这200个图像块中选出排名靠前的40个图像块作为ROIs;这样一来,就可以确保没有代表性的图像块属于空白区域,过度染色的图像块被忽略。平均而言,这40个ROIs代表了整个病理图像组织区域的15%。接下来,我们使用SqueezeNet来对这40个ROIs进行计算,将最后一层替换为512维向量。见下图:
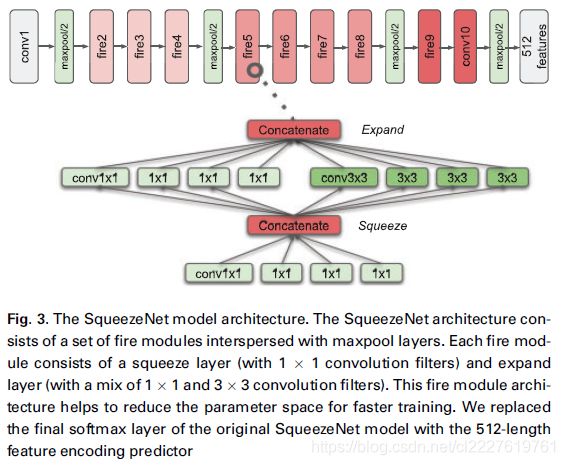
使用相似性损失和Cox损失对模型进行优化。由于SqueezeNet运算高效,所以我们可以在对大量图像块进行训练的同时又不损失性能。我们使用验证集对模型进行调优,选取验证集上最优的模型。为了评估模型的表现,我们使用测试集数据计算C-index。
Multimodal dropout
Dropout是深度学习中经常采用的一种正则化技术,训练过程中随机失活一部分神经元,迫使其他神经元参与进来,为失活的神经元做出预测。这一技术减轻了模型的过拟合,增强了模型的泛化能力。在本研究中,我们设计了dropout的一个变种,称为multimodal dropout,用它来提高模型应对缺失值的能力。在multimodal dropout中,我们不去失活神经元,我们要做的是丢弃某个模态数据的全部特征向量,同时增加其他模态数据的权重。在训练的过程中,对于每一种模态的数据,丢失的概率是 P P P,迫使模型创建的特征表示能够对于缺失数据具有鲁棒性。经过实验,我们确定最佳的 P P P等于25%。
可视化
T-SNE方法经常被用来将高维数据映射到低维空间。与主成分分析等其他的降维方式不同的是,T-SNE通过将向量相似性转变为联合概率,产生视觉上可分的聚类簇,能够代表数据中的特征模式。本文中,我们使用T-SNE进行聚类,每个患者都有一个512维的向量,我们通过聚类对它们之间的关系进行可视化。因为T-SNE的运算比较费时,我们先用PCA将512维向量降维到50维,然后再使用T-SNE将50维降到2维。
结果和讨论
无监督特征学习的结果
我们首先对无监督学习所编码的泛癌特征进行可视化。具有相似特征表示的一群患者往往拥有相同的种族、性别和肿瘤类型,尽管模型没有使用这些变量对模型进行训练。

这表明神经网络模型以一种无监督的方式学习到了不同模态数据之间的联系。这些结果表明我们的无监督模型能够有效地对多模态数据进行概括和总结,其编码的信息可以被当做“patient profile”。
multimodal dropout的评估结果
接下来,我们看一下使用了multimodal dropout对结果的影响。我们将模型训练了80个epoch,结果如下图:

上图表明训练过程中使用multimodal dropout可以提升验证集的C-index,这表明multimodal dropout可以有效应对缺失值带来的影响。
泛癌预后预测的结果
接下来,我们使用训练好的模型在测试集上预测单一类型肿瘤的预后以及泛癌的预后。我们比较了不同模态数据的组合的结果,每种组合都包含临床数据,我们还评估了multimodal dropout在这些组合中的影响。我们发现只有在临床信息和mRNA进行组合的时候,multimodal dropout没有提高效果。对于组合了所有模态数据的模型,15/20类型的肿瘤使用multimodal dropout的时候都有C-index的提高,平均提升为2.8%。使用较少的模态数据进行组合的时候,也可以得到类似的结果。在泛癌数据上,整合了所有模态数据以后,使用multimodal dropout的C-index为0.78,不使用multimodal dropout的C-index为0.75。
不同数据模态的效果
接下来,我们研究不同模态之间组合的结果,探讨基因组学和影像组学模态数据是否对预后预测很重要。我们发现miRNA是富含信息最多的模态,而mRNA含有的信息最少,对于baseline来说,泛癌的C-index为0.75 vs. 0.60。对于KICH,组合四种模态数据达到了0.95的C-index值。另外,对于其中六种肿瘤,整合了临床信息、miRNA和数字病理图像所得到的结果和整合四种模态的数据所得到的结果基本一致,这说明在这些类型的肿瘤中,mRNA对于预后预测没有那么重要。比如,对于KIRP,卵巢癌和肺腺癌来说,使用临床信息,miRNA和数字病理图像这三种模态的数据就足以预测预后了。
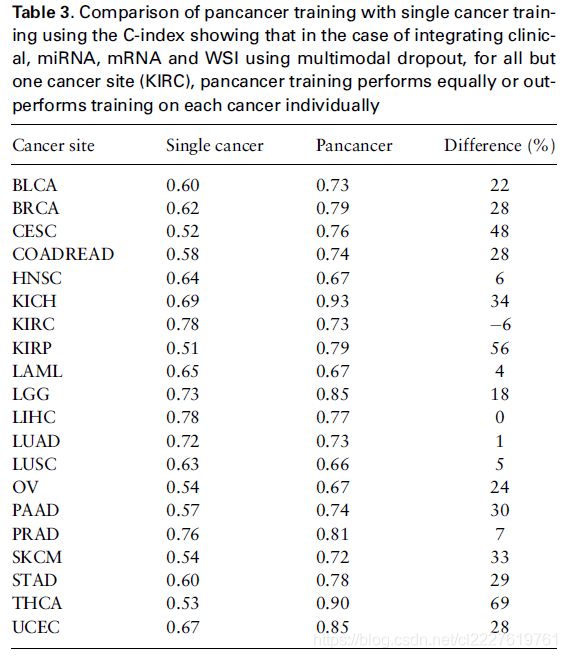
泛癌预训练得到的模型和单一肿瘤训练的模型的比较
接下来,我们测试由泛癌得到的模型在单一肿瘤上的效果与单一肿瘤训练的模型的效果进行比较。我们发现除了KIRC以外,由泛癌训练的模型在单一肿瘤上的效果均比单独训练单一肿瘤的效果更好。
和以前研究的比较
以前的研究都集中于某一种类型的肿瘤的预后预测。比如有研究在TCGA-KIRC上得到了0.77的C-index,我们的研究结果为0.74,比其稍差。然而,在肺腺癌上,我们的模型取得了0.726的C-index,这要比有研究得到的0.691的C-index高。总体而言,我们无法“完美”地比较我们的结果和以往研究的结果,因为以往研究中,常常将缺失值进行了剔除,但是我们的研究可以很好地将这些有缺失的数据利用上;此外,我们的研究在20种类型的泛癌数据上进行了训练,得到了达到甚至超过以往研究的结果。
结论
在本研究中,我们提出了一种多模态数据融合的方法来预测肿瘤预后。首先,我们使用无监督的方式对患者的多模态数据进行了编码,编码为一种独立于数据类型和模态的特征表示。然后,我们对这些特征在特征空间中进行了可视化。这些特征集成了患者的多模态数据,使得我们可以利用机器学习方法对这些"patient profile"进行系统的比较和分析。因此,这些编码特征在很多领域都可以被使用,比如预后预测和治疗方案的推荐。
然后我们使用这些特征表示对单一肿瘤和泛癌的预后进行了预测。在20种肿瘤上,我们的模型达到了平均0.784的C-index。而且,我们的模型以无监督的方式,能够在充分利用缺失数据的情况下,在KICH上达到非常高的准确率。
我们的模型在很多方面都有优点。我们展示了如何建立泛癌预测模型,另外,我们展示了如何进行多模态数据融合,并且提出了一种新的dropout方法,能够在某个模态数据缺失的情况下,充分利用该样本。具体来说,在以一种无监督的方式学习不同模态数据之间关系的同时,我们的模型能够为每个患者生成独特的特征表示。最后,我们提出一种有效的数字病理图像分析方法,以采样的方式得到的ROIs能够代表患者肿瘤区域的15%。
展望
尽管我们提出的方法可以从WSI中选择图像块,但是仍有提升空间。另外改善用于编码特征的网络结构对于提升模型表现也非常重要。将来我们的研究将不再随机选择图像块,而是关注那些重要的图像块。另外,我们将使用更深的网络结构以及数据增强操作。此外,融合更多模态的数据也是我们将来要做的工作,在本研究中,由于资源的限制,我们没能利用TCGA数据库中的DNA甲基化数据,DNA拷贝数数据等,这些可能都和预后有关。