论文:A Deep Generative Model for Molecule Optimization via One FragmentModification
A Deep Generative Model for Molecule Optimization via One FragmentModification
利用变分自编码器的生成模型
训练:输入两个对比的分子,生成断点,需要删除增加的片段
生成:从隐向量空间中采样 z , 和Mx一起计算出断点,和需要删除或添加的片段。
We first quantified the difference between Mx and My using the optimal graph edit distance between their junction tree representations Tx and Ty, and derived the optimal edit paths to transform Tx to Ty.
我们首先使用Mx和My的连接树表示Tx和Ty之间的最佳图形编辑距离来量化Mx和My之间的差异,并推导出将Tx转换为Ty的最佳编辑路径。然后导出将转成的最优编辑路径,该量化还在图比较期间确定了Mx上的断开点
我们使用2048维半径为2的二元摩根指纹来表示分子,并使用谷本系数来测量分子相似性
在这里,该团队提出了用于分子优化的新型生成模型,该模型更近似于硅化学修饰。该方法被称为「带有一个片段的修饰符」或 Modof。遵循基于片段的药物设计理念,Modof 预测分子上的单个断开位点,并通过改变该位点的片段(例如,环系统、接头和侧链)来修饰分子。
与现有的对整个分子图进行编码和解码的分子优化方法不同,Modof 从一个断开位置优化前后分子之间的差异中学习和编码。为了修改一个分子,Modof 只生成一个片段,该片段通过解码从潜在“差异”空间中抽取的样本来实例化预期的差异。然后,Modof 在断开连接的站点移除原始片段,并在该站点附加生成的片段。

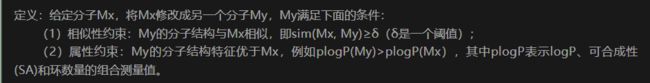
相似性约束和属性约束
定义:给定分子Mx,将Mx修改成另一个分子My,My满足下面的条件:
(1)相似性约束:My的分子结构与Mx相似,即sim(Mx, My)≥δ(δ是一个阈值);
(2)属性约束:My的分子结构特征优于Mx,例如plogP(My)>plogP(Mx),其中plogP表示logP、可合成性(SA)和环数量的组合测量值。
logP 是预测值 r2=0.918
参考文献 21 22
通过 Wildman 和 Crippen 方法估计 Crippen logP
使用预定义片段的评分函数计算合成可访问性,使用支持向量机分类器预测 DRD2 属性
利用非线性分类器结合分子性质的多个期望函数对量子电动力学性质进行了预测
我们在 Modof 中使用了一对分子(Mx,My)作为训练实例,其中 Mx 和 My 同时满足相似性和属性约束,而 My 与 Mxin 的不同之处仅在于一个断开位点的一个片段。 我们构建了这样的训练实例如下。 我们首先使用它们的连接树表示 Tx 和 Ty 之间的最佳图形编辑距离 26 来量化 Mx 和 My 之间的差异,并得出转换 TxtoTy 的最佳编辑路径。 这种量化还在其图形比较过程中确定了 Mx 的断开位置。 有关此过程的详细信息,请参见第 S4 节。 将满足相似性和性质约束且仅具有一个断开位置的已识别分子对用作训练实例。 对于具有高度相似性的一对分子(例如,高于 0.6),它们很可能只有一个断开位点,如第 S5 节所示。(每对分子相似,而且一个属性优于另一个,且仅具有一个断开位置)
分子相似度计算我们使用2048维半径为2的二元摩根指纹来表示分子,并使用谷本系数来测量分子相似度。
方法:
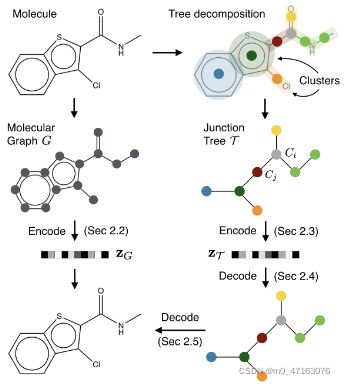
Modof 一次修饰分子的一个片段(例如,环系统、接头、侧链),因此只对需要修饰的片段进行编码和解码。 片段被修改的 M 的位点称为断开位点并表示为 nd,它对应于连接树表示中的一个节点(在“分子表示和符号”中讨论)。 图 3 展示了 Modof 的概述。 所有算法都在第 S14 节中介绍。 S5 节介绍了关于单断开站点基本原理的讨论。
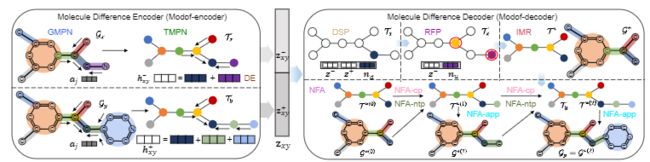
Fig. 3|Modof Model Overvie
分子表示和符号


Modof-encoder

1,图的消息传递机制

这种消息传递机制没见过,好复杂
2,树的消息传递机制

3. Difference Embedding (DE)

Molecular Difference Decoder (Modof-decoder)

1,断点识别 Disconnection Site Prediction (DSP)

2,移除片段预测

3

4


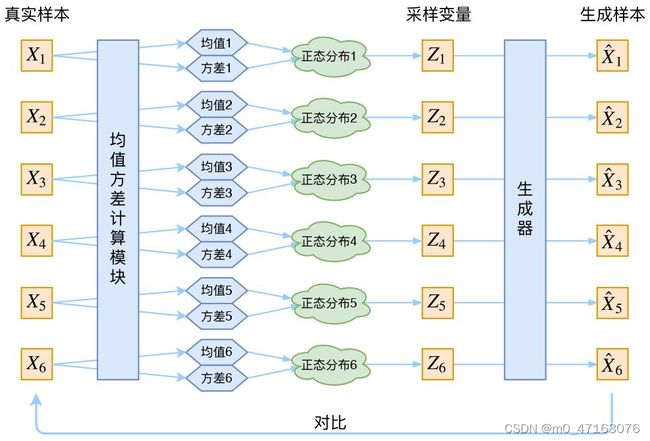
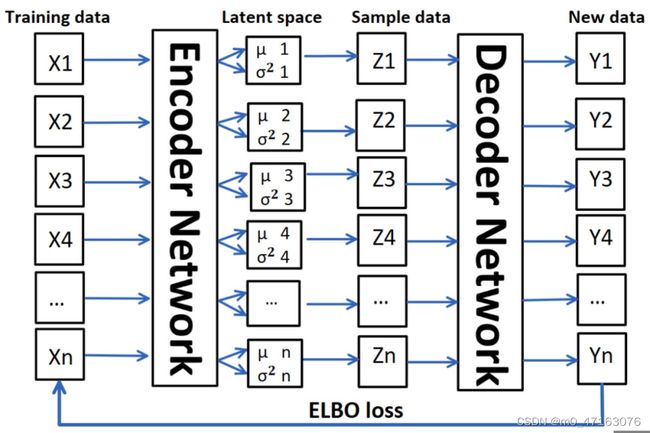
补充:先验分布、后验分布、似然估计这几个概念是什么意思,它们之间的关系是什么?
变分自编码器看来是搞不懂了,留个图,粗略理解一下
相关论文:Junction Tree Variational Autoencoder for Molecular Graph Generation
JTVAE 已经被封装到包里面了,可以直接使用,教程
我们的成分词汇表,例如环、键和单个原子,被选择得足够大,以便给定分子可以被重叠的成分或原子簇覆盖。 簇的作用类似于图形模型中的团,因为它们具有足够的表现力,以至于分子可以被重叠的簇覆盖而不会形成簇循环。 从这个意义上说,这些簇在分子图的(非最佳)三角剖分中充当了派系。 我们形成这样的簇的连接树,并使用它作为分子的树表示。 由于我们对团的选择是先验的,因此我们不能保证对于任意分子存在具有此类簇的连接树。 但是,我们的集群是建立在训练集中的分子的基础上的,以确保可以找到相应的连接树。 根据经验,我们的集群覆盖了测试集中的大部分分子
2.1 连接树
树分解通过将某些顶点收缩到单个节点来将图G映射到连接树,从而使G成为无循环的

将诱导子图视为集群标签,连接树是带有标签词汇表X 的标签树,通过我们的分子树分解,X只包含环和单边。因此标签词汇表X 是有限的(对于一个有250K分子的标准数据集,|X|= 780)。
我们的集群词汇 X 包括化学结构,例如键和环(图 3)
分子的树分解在这里,我们展示了为分子量身定制的树分解算法,它在化学中找到了根源(Rarey & Dixon,1998)。 我们的集群词汇 X 包括化学结构,例如键和环(图 3)。
给定一个图g,我们首先找到它所有的简单环,以及它的边不属于任何环。如果两个简单的环有两个以上重叠的原子,它们就会合并在一起,因为它们构成了一个特定的结构——回忆起桥联化合物(Clayden et al., 2001)。每一个环或边都被认为是一个簇。然后,通过在所有相交的聚类之间添加边来构造聚类图。最后,我们选择它的一个生成树作为g的结树(图3)。由于环合并,结树中的任意两个簇最多有两个相同的原子,有利于图解码阶段的高效推理。具体操作步骤请参见附录
树编码
我们类似地使用树消息传递网络对 TG 进行编码。 每个cluster Ci 由代表其标签类型的单热编码xi 表示。 每条边(Ci,Cj)都与两个消息向量mij 和mji 相关联。 我们选择任意叶节点作为根,并分两个阶段传播消息。 在第一个自底向上阶段,消息从叶节点开始并迭代地传播到根节点。 在自顶向下阶段,消息从根传播到所有叶节点。 mij 的消息被更新为:
![]()
(mij 是什么?文章中好像没有定义呀)

消息传递遵循 miji 仅在其所有前体 {mki|k∈N(i)\j} 已被计算时才计算的时间表。 这种架构设计的动机是基于树的信念传播算法,因此与图编码器不同。
在消息传递之后,我们通过聚合其内部消息获得每个节点的潜在表示
![]()
最终的树表示 是 hTG=hroot,它编码了一个有根树(T,root)。 与图编码器不同,我们不应用节点平均池化,因为它会混淆树解码器首先生成哪个节点。zTG 的采样方式与图编码器中类似。 为简单起见,我们从现在开始缩写为 zTG 为 zT
这个树形编码器在我们的框架中扮演着两个角色。 首先,它用于计算zT,它只需要网络的自底向上阶段。 其次,在从 zT 解码树之后,它被用于计算 messageŝ mij over the entirê T,以在图解码期间提供每个节点的基本上下文。 这需要自上而下和自下而上的阶段。 我们将在2.5部分详细说明
2.4. Tree Decoder
我们用一个树型结构的解码器将一个结树从它的编码中解码出来。树是按自顶向下的方式构造的,每次生成一个节点。如图4所示,我们的树解码器从根遍历整个树,并按深度优先顺序生成节点。对于每个访问的节点,解码器首先进行拓扑预测:该节点是否有子节点要生成。当一个新的子节点被创建时,我们预测它的标签并递归这个过程。回想一下,簇标签表示分子中的子图。当阳极没有更多的子节点时,解码器回溯
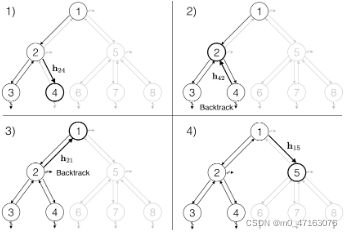
在每个时间步,一个节点从当前树中的其他节点接收信息以进行这些预测。 当树被增量构建时,信息通过消息向量传播。 形式上,设̃E={(i1,j1),···,(im,jm)}是在深度第一次遍历中遍历的边T= (V,E),其中m= 2|E|每条边都在 方向。 该模型访问它的节点时间。 让 ̃Et 成为 ̃E 中的第一个边。 Themesagehit,jtis 通过以前的消息更新:
![]()
拓扑预测:当模型访问节点时,它对它是否还有要生成的孩子进行二元预测。 我们通过结合zT, node features xit and inward messages hk,it 计算这个概率 ,通过一个隐藏层网络,后跟一个 sigmoid 函数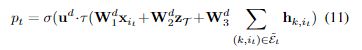

3,Experiments
我们的评估工作衡量了分子生成的各个方面。 前两个评估遵循先前提出的任务(Kusner 等人,2017)。
我们还介绍了第三个任务——约束分子优化Molecule reconstruction and validity (分子重建和有效性)We test the VAEmodels
on the task of reconstructing input molecules fromtheir latent
representations, and decoding valid moleculeswhen sampling from prior
distribution. (Section 3.1)贝叶斯优化 除了生成有效分子之外,我们还测试了该模型如何生成具有所需特性的新型分子。
为此,我们在潜在空间中执行贝叶斯优化以搜索具有指定属性的分子. (Section 3.2)Constrained 分子优化 其任务是修改给定的分子,以改善特定的性能,同时约束偏离原始分子的程度。在药物发现中,这是一个更现实的场景,新药物的开发通常从已知分子(如现有药物)开始(Besnard et al.,2012)。由于这是一个新任务,我们不能与任何现有的基线进行比较. (Section 3.3)
下面我们描述了跨任务共享的数据、基线和模型配置。 特定于任务的部分提供了其他设置详细信息。
数据我们使用来自 Kusner 等人(2017)的 ZINC 分子数据集进行实验,具有相同的训练/测试拆分。 它包含从 ZINC 数据库中提取的大约 25 万个药物分子(Sterling & Irwin,2015)
基线 我们将我们的方法与基于 SMILES 的基线进行比较:1) Character VAE (CVAE) (G ́omez-Bombarelliet al., 2016),它逐个字符地生成 SMILES 字符串; 2) 语法 VAE (GVAE) (Kusner et al., 2017),根据上下文无关语法给出的句法约束生成 SMILES; 3) 语法导向的 VAE (SD-VAE) (Dai et al., 2018),通过属性语法结合了 SMILES 的语法和语义约束。 对于分子生成任务,我们还比较了直接生成图的原子标签和邻接矩阵的 GraphVAE (Simonovsky & Komodakis, 2018)
模型配置 为了与上述基线相比较,我们将潜在空间维数设为56,即树型表示和图型表示htandhg28维。完整的培训细节和模型配置在附录中提供
3.1. Molecule Reconstruction and Validity
3.2. Bayesian Optimization
3.3. Constrained Optimization
第三个任务是在受限场景中执行分子优化。 给定一个分子m,任务是找到一个不同的分子m',它具有最高的属性值,分子相似度sim(m,m')≥δ对于某个阈值δ。 我们使用与 Morgan fingerprint (Rogers & Hahn, 2010) 的 Tanimoto 相似性作为相似性度量,并使用惩罚 logP 系数作为我们的目标化学性质。 对于这个任务,我们用 JT-VAE topredicty(m) 从 m 的潜在嵌入中联合训练一个属性预测器 F(由前馈网络参数化)。 为了优化分子,我们从其潜在表示开始,并在潜在空间中应用梯度上升来提高预测得分 F(·),类似于 (Mueller et al., 2017)。 应用 K= 80 梯度步骤后,K 分子从产生的潜在轨迹中解码,我们报告满足相似性约束的具有最高 F(·) 的分子。**如果解码的分子之一满足约束并且与原始分子不同,则修改成功**
**分子相似度sim(m,m')≥δ对于某个阈值δ**
**使用与 Morgan fingerprint (Rogers & Hahn, 2010) 的 Tanimoto 相似性作为相似性度量**
**使用惩罚 logP 系数作为我们的目标化学性质**