【学习笔记】山东大学生物信息学-04 蛋白质结构预测与分析
课程地址:山东大学生物信息学
文章目录
- 四、蛋白质结构预测与分析
-
- 4.1 蛋白质的二级结构
- 4.2 蛋白质的三级结构
- 4.3 三级结构可视化软件 VMD
- 4.4 计算方法预测三级结构
- 4.5 三级结构的比对
- 4.6 蛋白质分子表面性质
- 4.7 获取蛋白质四级结构
- 4.8 蛋白质-蛋白质分子对接
- 4.9 蛋白质-小分子分子对接
- 4.10 虚拟筛选与反向对接
- 4.11 分子动力学模拟
四、蛋白质结构预测与分析
4.1 蛋白质的二级结构
常见的二级结构单元
-
详见视频:蛋白质的二级结构-01 P73
-
螺旋:最常见的就是 α 螺旋。还有三转角螺旋、五转角螺旋等。
-
β 折叠 (
βsheet) : β 折叠由 β 折片 (β-strand) 平行排列而成。序列上可能相隔很远,但空间上并排在一起,彼此间形成氢键。 -
无规卷曲 (
coil): 无规律松散结构。 -
β 转角 (
turn) : 如果肽链发生了急转弯(角度大于 90°),这个转弯结构叫 β 转角。

-
蛋白质的二级结构常用图形或字母形式来描述:
H:螺旋
E:β 折叠
T:代表转角
空白:松散的 coil 结构

-
DSSP (Definition of Secondary Structure of Proteins),即,蛋白质二级结构定义词典。DSSP 并不预测二级结构,而是根据二级结构的定义对已经测定三级结构的蛋白质的各个位置指认出是哪种二级结构。
DSSP 网址:http://swift.cmbi.ru.nl/gv/dssp
-
直接下载 PDB 中已有结构的 DSSP 文件:
http://www.pdb.org/pdb/files/3cig.dssp (替换结构名 3cig 即可)
ftp://ftp.cmbi.ru.n/pub/molbioldata/dssp/3cig.dssp

从 PDB 获取蛋白质二级结构信息
- 详见视频:蛋白质的二级结构-02 PDB P74
- PDB 数据库里存储的所有蛋白质的一级结构和二级结构都以 FASTA 的格式存储在一个叫做 “ss.txt” 的文本文件里(文件很大,使用不方便)。
http://www.rcsb.org/pdb/files/ss.txt
http://www.rcsb.org/pdb/files/ss.txt.gz (压缩文件 30.6M) - 自编程序 Biotools(打不开)http://1.51.215.28/~gongj/biotools/
输入 PDB ID 自动从网上获取 dssp 文件并抽取出一级和二级结构的序列信息。
预测蛋白质二级结构
- 详见视频:蛋白质的二级结构-03 PDB P75
- 对于未知结构的蛋白质,可以通过氨基酸序列,预测其二级结构。目前的二级结构预测软件只预测 α 螺旋和 β 折叠,不预测其他二级结构单元。
- 常用软件:
PSIPRED http://bioinf.cs.ucl.ac.uk/psipred
Jpred3 http://www.compbio.dundee.ac.uk/www-jpred/
PREDICTPROTEIN http://www.predictprotein.org/
SSpro http://scratch.proteomics.ics.uci.edu/
PSSpred http://zhanglab.ccmb.med.umich.edu/PSSpred/
PREDATOR http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms::predator
GOR V http://gor.bb.iastate.edu/
4.2 蛋白质的三级结构
- 三级结构是指整条多肽链的三维空间结构,即,包括骨架和侧链在内的所有原子的空间排列。
- 测定三级结构的方法:X 射线衍射法(X-ray Crystallography)、核磁共振法(NMR,Nuclear Magnetic Resonance)、冷冻电子显微镜技术等。
- PDB 中绝大多数蛋白质的三级结构是用 X 射线衍射法测定的。
- 无法结晶的蛋白质可以利用核磁共振法在液体环境中测定(只能用于测定质量小于 70kD 的分子,约 200 多个氨基酸的蛋白质)。
- 详见视频:蛋白质的三级结构 PDB P76
- PDB 还可以通过序列相似性搜索获得与输入序列同源的蛋白质的三级结构。

- 下载 PDB 文件,用于后续三维可视化软件读取。

4.3 三级结构可视化软件 VMD
- 详见视频:三级结构可视化软件 VMD-01 鼠标操作 P77
- VMD http://www.ks.uiuc.edu/Research/vmd/ 免费,需要注册

- 详见视频:三级结构可视化软件 VMD-02 外观设置 P78


- 详见视频:三级结构可视化软件 VMD-03 设置多个外观状态叠加 P79

- 详见视频:三级结构可视化软件 VMD-04 外观设置 P80

4.4 计算方法预测三级结构
- 常用方法:
- 从头计算法 ab initio
- 同源建模法 homolog modeling (首选)
- 穿线法 threading
- 综合法 ensemble method
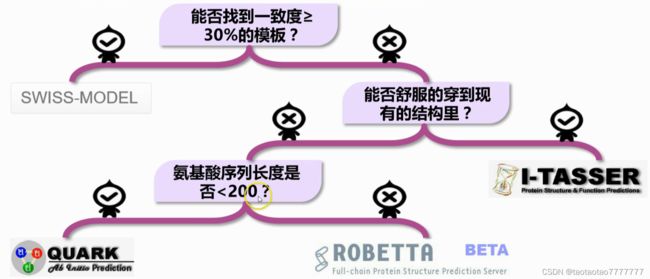
同源建模法 SWISS-MODEL
- 原理:相似的氨基酸序列对应着相似的蛋白质结构。
- 步骤流程:
- 找到与目标序列同源的已知结构作为模版(目标序列与模版序列间的一致度要≥30%)。
- 为目标序列与模版序列(可以多条)创建序列比对。通常比对软件自动创建的序列比对还需要进一步人工校正。
- 根据第二步创建的序列比对,用同源建模软件预测结构模型。
- 评估模型质量并根据评估结果重复以上过程,直至模型质量合格。

- SWISS-MODEL 是一款用同源建模法预测蛋白质三级结构的全自动在线软件。
- 详见视频:计算方法预测三级结构-02 SWISS-MODEL P82


- 预测效果:如果目标序列与模板序列一致度极高,那么同源建模法是最准确的方法。

◆ 如果目标序列与模板序列之间的一致度 <30%,那么同源建模法是不适用的。
◆ 一致度达到 30%,模型的准确度就能达到 80%,模型可以用来寻找功能位点以及推测功能关系等。
◆ 一致度达到 50%,模型的准确度就能达到 95%,可以根据模型设计定点突变实验,甚至做晶体结构置换,辅助完成真实结构的测定。
◆ 一致度达到 70%以上,可以认为预测模型完全代表真实结构,可以用来进行虚拟筛选、分子对接、药物设计等结构功能研究。
◆ 特例情况:虽然序列一致度达到很高水平,但是结构却并不相同。
如下图,晶体结构发生了一个氨基酸的错位,导致最后一个折片的方向发生 60°的扭转,使得 C 端残基的空间位置发生改变,并因此导致两者功能的差异。

穿线法 I-TASSER
-
原理:不相似的氨基酸序列也可以对应着相似的蛋白质结构。(同源建模法找不到相似模板时使用)
-
已知的蛋白质结构约 10 万个,其所具有的不同的结构拓扑只有 1393 个,且自 2008 年就没再有新的结构拓扑产生。
-
把目标序列像线一样穿到目前现有的结构里,穿到哪个结构最舒服,就用哪个结构作为预测模板;并通过能量方程,穿得舒服,能量就低,根据最舒服的穿法构建最终模型。

-
这种方法计算量大,耗时。
-
I-TASSER 是一款用穿线法预测蛋白质三级结构的在线软件,在连续几届蛋白质结构预测比赛中皆排名第一。作者为美国密歇根大学的张阳教授。
-
详见视频:计算方法预测三级结构-03 穿线法 I-TASSER P83



- 通过任务号查找任务


- 预测结果


- 预测模型信息:
(1) 模型质量评估系数C-score: [-5, 2] 分值越高模型可信度越高;
(2)TM-socre: 两两结构相似度系数,> 0.5说明模型具有正确的结构拓扑,可信;<0.17说明模型属于随机模型,不可信;
(3)RMSD: 两两结构间的距离偏差。
- 预测出的蛋白质功能,以及有可能与之结合的配体和该配体的结合位点。
- 通过任务号查找任务
从头计算法:QUARK
- 原理:1973 年《科学》Anfinsen : 蛋白质的三维结构决定于自身的氨基酸序列,并且处于最低自由能状态。
- 有些特殊蛋白质用穿线法也预测不了三级结构模型,此时可以用从头计算法,模拟一个肽段在三维空间内所有可能存在的姿态,并计算每个姿态的自由能,最后将==自由能最低的姿态作为预测结果==(计算量大,轻易不使用)。
- QUARK 是一款用从头计算法预测蛋白质三级结构的在线软件,适用于没有同源模板的蛋白质,且氨基酸序列长度 200 以内,计算时间约 2 天以上。
- 详见视频:计算方法预测三级结构-04 从头计算法 QUARK P84
- 根据 TM-score 查看模型是否合格可用。

综合法 ROBETTA
- 原理:综合了同源建模法、穿线法和从头计算法等多种方法,将氨基酸序列分段,情况不同的片段采用不同的方法预测。
- ROBETTA 综合了同源建模法和从头计算法两种方法。能找到模板的区域用同源建模法,找不到的区域用从头计算法。(所需时间是以上方法中最长的,要排队)
- 详见视频:计算方法预测三级结构-05 综合法 ROBETTA P85
- 整条序列被分成多个 domains , 每个 domain 采用不同的方法分别预测。同源建模法需要几个小时到几天的时间,从头计算法需要几天到几周的时间,这取决于目标序列的预测“难易”程度。

- 总结:到底用哪种方法?
模型质量评估
- 模型质量评估软件 (Model Quality Assessment programs,
MQAPs)
对于通过计算方法获得的模型,必须进行必要的模型质量评估,以确定模型的可靠性。模型质量评估软件并不比较预测模型跟真实结构的差别大小,而是从空间几何学、立体化学和能量分布三方面评估一个模型的自身合理性。 - 大多软件自带模型质量评估系数:
◆ I-TASSER:C-score的取值范围是 [-5,2] , 分值越高模型越可靠。
◆ Swiss-Model:QMEAN4的取值范围是 [0,1] , 分值越高模型越可靠。
◆ QUARK:TM-score>0.5 说明模型的可靠,<0.17 说明模型不可信。

- 至少需要 3 个模型质量评估体系认为模型可靠,才能认为模型可靠。因此需要借助第三方模型评估软件。
- 详见视频:计算方法预测三级结构-06 模型质量评估 P86
- 常用 SAVES 提供 6 个模型质量评估软件。可以一次 6 个软件一起作,也可以单独选择常用的
Verify3D、PROCHECK和ERRAT。


- ProQ 通过 LGscore 和 MaxSub 两个值反应模型质量。计算时间只有十几秒,并且评估结果明确。

- ModFold 评估结果明确。评估需要 30 分钟左右,并且一个 email 地址一次只能提交一个评估任务。
4.5 三级结构的比对
- 结构比对就是对蛋白质三维空间结构的相似性进行比较,他是蛋白质结构分析的重要手段之一。
- 可用于探索蛋白质进化及同源关系
- 改进序列比对的精度
- 改进蛋白质结构预测工具
- 为蛋白质结构分类提供依据
- 帮助了解蛋白质功能
- 结构比对的结果可以用很多种参数来衡量,最常用的是 root mean squared deviations (
RMSD)。
如果两个结构的 RMSD 为 0 埃,那么它们结构一致,可以完全重合;一般来说,RMSD 小于 3 埃时,认为两个结构相似。
SuperPose
- SuperPose 蛋白质结构叠合软件,可以将两种结构叠合在一起,并给出两者的 RMSD。
- 详见视频:三级结构的比对-01 SuperPose P87


SPDBV 选择叠合
- SPDBV 是一款蛋白质结构分析软件,也是一个蛋白质同源建模平台。其结构叠合功能十分出色。可进行整体智能叠合,或者选择性叠合。下载后无需安装,直接运行。
- 详见视频:三级结构的比对-02 SPDBV 选择叠合 P88
4.6 蛋白质分子表面性质
- 表面形状 ( VMD:
SURF representation) - 表面电荷分布 (
electrostatic potential) - 表面残基可溶性 (
solvent acessibiliy): 即残基与溶剂接触的程度,也就是哪些地方是掩埋在内部的 (buried) ; 哪些地方是露在表面的 (exposed) ; 哪些地方介于掩埋与暴露之间的中间状态 (intermediate)。
- 需要给 VMD 安装 APBS 插件。
- 详见视频:蛋白质分子表面性质-01 VMD-APBS P89
4.7 获取蛋白质四级结构
-
蛋白质四级结构是独立的三级结构单元聚集形成的复合物,其中每个独立三级结构称为亚基,也称为单体 (
monomer)。含两个亚基的蛋白质称为二聚体 (dimer); 含三个亚基则称三聚体 (trimer) ; 还有四聚体 (tetramer); 五聚体 (pentamer); 六聚体 (hexamer) 等。 -
蛋白质异常聚集导致的疾病
◆ 诱发神经系统退行性病变的淀粉样蛋白 (amyloid-protein, A) 是蛋白质序列相同但四级结构不同而诱发疾病的典型代表。
◆ 阿尔茨海默病 (Alzheimer disease,AD) : 在 AD 发生过程中出现淀粉样蛋白。
◆ A 是由特殊水解酶对其前体蛋白的水解作用产生的。
◆ A 有两种构象,一种为螺旋且可溶而存在于健康个体脑组织,此类 A 为单体没有四级结构;另一种为片层且是多个 A 聚集形成的链间片层,此类 A 不溶且出现在 AD 患者脑组织。
◆ 诱发 A 从可溶螺旋转变成不溶片层聚集体的机制不清,但已广泛被证实这种构象转变是 AD 的重要诱因。 -
四级结构的获取:实验方法(高成本)
(1) X 射线衍射法 X-ray Crystallgraphy:可以精确获得复合体结构,但技术难度较大,因为复合体很难获取并成功结晶。
(2)冷冻电子显微镜技术 Cryoclectron Microscopy:不能获得精确的结构,只能获得影子或外形轮廓,之后再根据已有的同源蛋白质的晶体结构对影子中的单体进行同源建模,再把模型套到影子里。 -
四级结构的获取:数据库获取(蛋白质相互作用关系数据库)
◆ DIP (the Database of Interacting Proteins): 实验方法测定的蛋白质之间的相至作用。http://dip.doe-mbi.ucla.edu/dip/Main.cgi
◆ BioGRID (the Biological General Repository for Interaction Datasets): 主要收集模式生物物种中涉及的蛋白质间相互作用,是各种相互作用的数据集。http://thebiogrid.org/
◆ STRING 实验测定已知的及计算方法预测的蛋白质间相互作用。http://string-db.org/
4.8 蛋白质-蛋白质分子对接
- 分子对接 (
docking): 蛋白质-蛋白质分子对接。(用计算方法预测蛋白质的四级结构) - 目前做分子对接的软件都是尝试所有可能的结合形式,并根据打分函数(能量的高低)给每种形式打分排名。
- 对接的过程中会考虑如下因素:
◆ 形状互补
◆ 亲疏水性
◆ 表面电荷分布 - 两种蛋白质-蛋白质分子对接:
Rigid Docking刚性对接:目前可用的大多数软件为刚性对接。
Flexible Docking柔性对接:计算量大,可用软件少,且多为收费软件。 - 蛋白质相互作用常用对接软件
◆ ZDOCK: http://zdock.umassmed.edu/
◆ GRAMM-X: http://vakser.bioinformatics.ku.edu/resources/gramm/grammx
◆ 输出值都为多个对接状态(根据能量高低排序,能量低的排名靠前)。结果可用 VMD 查看。
◆ GRAMM-X 的输出结果,即多个对接状态都保存在同一个 PDB 文件中。该 PDB 文件包含多个 Frames,每个 Frame 为一个对接状态。保存输出结果中的某一个状态:在 VMD main 中选中当前的文件,右键 Delete Frames, 删去不要的那些状态,再右键 Save Coordinates,保存唯一留下的没删的状态。 - 详见视频:蛋白质相互作用常用对接软件 P92

- PDBePISAs 相互作用面分析
- 详见视频:PDBePISAs 相互作用面分析 P93

4.9 蛋白质-小分子分子对接
- Rigid Docking 刚性对接:小分子总是柔性的,蛋白质上结合小分子的部位被认为是刚性的。
- Flexible Docking 柔性对接:小分子总是柔性的,蛋白质上结合小分子的部位被认为是柔性的。
- AutoDock http://autodock.scripps.edu/ 需要先安装 python 2.5.4(高版本不兼容)
- 详见视频:蛋白质-小分子分子对接 AutoDock 安装 P94

- 详见视频:蛋白质-小分子分子对接 AutoDock 使用 P95-96
4.10 虚拟筛选与反向对接
虛拟筛选
- 虛拟筛选 Virtual screening (
VS) ,也称计算机筛选,即在进行生物活性筛选之前,在计算机上对化合物分子进行预筛选,以降低实际筛选化合物的数目,同时提高先导化合物的发现效率。(即,预筛选与蛋白质相互作用的小分子,挑虚拟筛选结果中结合得最好的小分子做实验验证。) - ZINC 数据库:化合物小分子数据库 http://zinc.docking.org
- 虛拟筛选过程:
- 从小分子数据库 ZINC 下载小分子 3D 结构;
- 程序处理下载的小分子文件;
- 软件处理蛋白质分子,设定对接空间范围;
- AutoDock Vina 程序批量完成对接;
- 对接结果分析,筛选出适合的小分子:按自由能高低排序,低的在前。
- 软件分析排名第一的小分子对接状态
- 从销售商处购买筛选出的小分子,实验验证。
- 用免费的 AutoDock Vina 做虛拟筛选
- 详见视频:虚拟筛选 P97
- 详见视频:AutoDock Vina 教程 P98
反向对接
-
反向对接 (
Target Fishing):是通过把一个小分子与多个靶标蛋白进行分子对接,寻找潜在的靶标。 -
当前软件和技术:还 没有标准和成熟的免费软件来实现,只有少数收费软件能实现此功能,以及很少的科研单位通过自己的算法和对已有对接程序的改造来实现。
-
靶标数据库 http://bioinfo-pharma.u-strasbg.fr/scPDB
-
scPDB: An Annotated Database of Druggable Binding Sites from the Protein DataBank
-
scPDB 收集了 PDB 数据库中有药物结合位点的蛋白。可根据配体、蛋白、结合方式为特征进行搜索。
-
当前版本包含:8077 entries, 2377 proteins and 5233 ligands 可以下载到本地使用,每个蛋白一个单独的文件夹,每个文件夹都包含如下文件:
ligand_xray.mol2: 从原始复合物中剥离的配体晶体结构
protein.mol2: 去除配体结构的蛋白受体结构
site.mol2: 去除配体结构的蛋白受体结构,仅包含结合位点 -
反向对接 (= 多个正向对接)
◆ 遍历蛋白数据库文件,获得每个蛋白质及其相关文件
◆ 软件自动探测结合位点,并标识结合区域自动循环,使小分子与每个蛋白受体对接
◆ 打分、筛选和排序
4.11 分子动力学模拟
- 分子动力学模拟 (Molecular Dynamic Simulation,MDS):用计算机来模拟原子及分子的物理运动过程。
- 软件:NAMD,CHARMM,DESMOND, GAUSS 等(需要超级计算机完成)。
- 详见视频:分子动力学模拟 P100