NBT|45种单细胞轨迹推断方法比较,110个实际数据集和229个合成数据集

摘要
轨迹推断(Trajectory Inference,TI),是分析从千上万单细胞的组学数据中推断细胞发育轨迹的重要方法,也被称为伪时序分析 (pseudotime analysis),该方法根据细胞表达模式的相似性对细胞进行排序。这为应用单细胞转录组学、蛋白质组学和表观组学数据研究细胞内的动态过程,如细胞周期、细胞分化和细胞激活等,提供了新的契机。
虽然轨迹推断工具已经有了七十多种,不过因为每个方法输入和输出模型差别很大,所以很难评判每个工具的性能。本篇文章通过对110个真实数据集和229个模拟数据集进行评估测试,分析比较了45种TI方法的细胞排序 (cellular ordering)结果、拓扑结构展示、扩展性和易用性。结果表明现有工具的互补性很好,各自有自己的适用范围,建议用户根据数据维度和轨迹拓扑结构挑选合适的方法。
由此本文总结了一套使用指南,可以帮助用户根据数据和分析需求选择最优方法。本文所有的测试数据和评估流程都可在https://benchmark.dynverse.org公开访问获得。该指南在帮助现有用户选择之余,对开发用于更大和更复杂的数据集的分析工具也有裨益。
黄金标准数据集和模拟数据集结果都有很好的代表性
110个实际单细胞转录组数据集和229个模拟单细胞转录组数据集:
模拟数据集: 采用多种数据模拟器包括基于热动力模型的调控网络模拟等生成的数据,模拟的数据参照了真实数据集的大小、差异表达基因数量和drop-out率和其他统计属性得到。模拟数据提供了精确的参考轨迹用于软件评估。
实际数据集:实际数来源于多种单细胞测序技术、多种样品和多个生物学过程;预期轨迹包含多种拓扑结构;如果实际数据集的发育轨迹是通过细胞分选或混合已知类型的细胞而不是单单依赖于表达量得来的定义为“黄金标准”数据 (有实验数据或先验知识支持),否则为“白银标准”。
在方法评估得分中,不同数据集的总体得分与包含金标准的真实数据集的得分具有中等至高度相关性(斯皮尔曼等级相关性在0.5-0.9之间),验证了黄金标准数据集和模拟数据集结果都有很好的代表性。
图解
评估流程的几个关键概念
下图 a展示了评估的流程、评判标准和结果存储; b每种方法预测的轨迹与参考轨迹在拓扑结构、分支分配、细胞排序的水平进行比较评估;c展示轨迹的多种拓扑结构,包括闭合环形、线性、单分叉、多分叉、树形、连接图(多种轨迹并存)、断开图 (多种不相连轨迹并存)。

d图更详细的解释了4种计算标准。HIM得分 (Hamming–Ipsen–Mikhailov)通过考虑边的长度和度分布评判预测的轨迹与参考轨迹的拓扑结构相似度; F1(branch)评判两个轨迹中最相似分支所含细胞的相似度;Cor(dist)通过计算细胞之间配对物理距离评估两个轨迹中的细胞排序一致性;Wcor(feature)评估两个轨迹鉴定的差异表达基因的一致性。

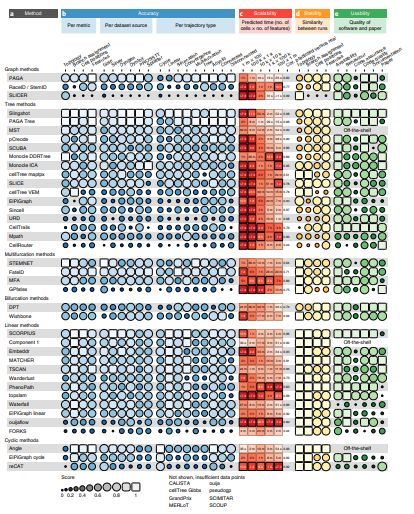
45种TI方法评估结果概览
45种TI方法根据它们所能推断的最复杂的拓扑结构分类,并展示了其是否依赖先验知识 (空白表示不需要,x是需要开始和终止细胞信息,x是需要细胞分组或time course信息)、Wrapper type (具体见https://github.com/dynverse/dynwrap)、运行平台、结果受算法或参数限制、可构建的拓扑轨迹类型、整体得分(准确性、拓展性、稳定性、易用性)。
整体来看得分最好的是PAGA, Slingshot, PAGA tree。线性拓扑方法中最好的是SCORPIUS。闭合环形拓扑中得分最好的是Angle。

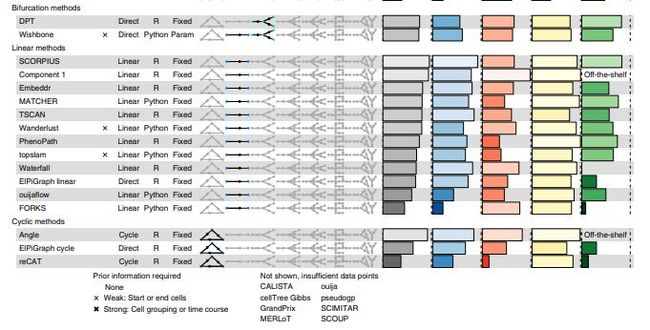
更详细的评估结果
热图展示了各个TI方法应用于不同的数据集、不同类型的拓扑轨迹时的准确性、多次运行时结果的稳定性、数据量增加时所需计算时间的变化、代码和文档的质量水平的得分。
不同的方法适用于不同的轨迹拓扑结构,而且受轨迹拓扑结构影响最大。
Slingshot应用于分析简单拓扑结构时效果最好,实际上它也倾向于预测对应数据的拓扑结构比较简单。PAGA,pCreode和RaceID/StemID在树形或更复杂的拓扑轨迹中效果更好。

可扩展性:随着需要处理的单细胞数据量和复杂度逐渐增加,各个工具的可扩展性也是评估的一个重要指标。
如图,列C是不同方法应用于不同数量的细胞和特征(no. of cells x no. of features)时所需的时间展示。颜色越深代表所需时间越长。k是thousands;m是million;cor是correlation,预测值和实际值的相关性值大小 (运行时间是数据集抽样时间的回归模型推测的)。
大部分方法都可以在12G内存的限制下工作,PAGA和STEMNET所需内存尤其少。
稳定性:两个相似的输入数据获得相似的轨迹预测结果。对一个数据集进行10次重抽样(抽取95%细胞,95%特征),评估同一个方法预测的轨迹的相似性,作为稳定性得分。
不同轨迹推断方法的互补关系
一种方法获得的结果准确性不高时,可以采用多种方法整合结果。下图展示了,采用1种、2种…多种方法组合后,获得最佳预测模型的可能性。从图中可以看出不同方法存在比较好的互补性,我们在实际分析时可以采用多种方法,整合分析结果。开发方法时,也可以做一个整合方法,优中选优。

用户指南 |关于方法选择
根据评估,方法的准确性主要取决于发育轨迹的拓扑结构,所以选择合适的轨迹推断方法也受用户的先验知识对自己研究的数据可能的拓扑结构的预期。用户可以根据这个指南图和先验拓扑轨迹知识,结合软件的准确性、可用性和所需时间综合评估选择。具体可以在线使用guidelines.dynverse.org,可以动态探索和查询评估结果。

几行代码运行所有的轨迹分析
应用不同方法的关键是统一输入和输出数据,这样用户就可以简单的同时运行多种TI方法,比较不同方法的预测结果并进行下游分析。比如仅需使用几行R代码(https://methods.dynverse.org)就可以绘制如下图形。
每种方法预测的轨迹投射到一个共有的空间纬度,通过计算每对预测模型的相似性,选择平均相似性得分最高的模型作为一致性模型。对于线性轨迹和二分轨迹 (bifurcating),方法已经比较成熟。而对树形结构、闭合环轨迹、断开图 (多种不相连轨迹并存)则还需要继续改善。

单细胞
收藏 北大生信平台” 单细胞分析、染色质分析” 视频和PPT分享
Science: 小鼠肾脏单细胞转录组+突变分析揭示肾病潜在的细胞靶标
Science:通过单细胞转录组测序揭示玉米减数分裂进程 | 很好的单细胞分析案例
Nature 首次对阿尔茨海默病进行单细胞转录组分析|详细解读
Cell 深度 一套普遍适用于各类单细胞测序数据集的锚定整合方案
骨髓基质在正常和白血病个体中的细胞图谱 Cell,Nature联袂解析
癌中之王:基质微环境塑造胰腺癌瘤内结构|Cell
Nature系列 整合单细胞转录组学和质谱流式确定类风湿性关节炎滑膜组织中的炎症细胞状态 详细解读
单细胞转录组教程汇总
10X单细胞测序分析软件:Cell ranger,从拆库到定量
Hemberg-lab单细胞转录组数据分析(一)- 引言
Hemberg-lab单细胞转录组数据分析(二)- 实验平台
Hemberg-lab单细胞转录组数据分析(三)- 原始数据质控
Hemberg-lab单细胞转录组数据分析(四)- 文库拆分和细胞鉴定
Hemberg-lab单细胞转录组数据分析(五)- STAR, Kallisto定量
Hemberg-lab单细胞转录组数据分析(六)- 构建表达矩阵,UMI介绍
Hemberg-lab单细胞转录组数据分析(七)- 导入10X和SmartSeq2数据Tabula Muris
Hemberg-lab单细胞转录组数据分析(八)- Scater包输入导入和存储
Hemberg-lab单细胞转录组数据分析(九)- Scater包单细胞过滤
Hemberg-lab单细胞转录组数据分析(十)- Scater基因评估和过滤
Hemberg-lab单细胞转录组数据分析(十一)- Scater单细胞表达谱PCA可视化
Hemberg-lab单细胞转录组数据分析(十二)- Scater单细胞表达谱tSNE可视化
如何火眼金睛鉴定那些单细胞转录组中的混杂因素
什么?你做的差异基因方法不合适?
单细胞分群后,怎么找到Marker基因定义每一类群?
在线平台如何做单细胞测序分析全套?有它so easy!
植物单细胞转录组的春天来了,还不上车?Science, PC, PP, MP, bioRxiv各一个
三人成虎,概率却不足十分之五?
一文掌握GSEA,超详细教程
这个只需一步就可做富集分析的网站还未发表就被CNS等引用超过350次
什么,你算出的P-value看上去像齐天大圣变的庙?
GO、GSEA富集分析一网打进
GSEA富集分析 - 界面操作
无需写代码的高颜值富集分析神器
去东方,最好用的在线GO富集分析工具
跨物种单细胞分析发现胰腺导管癌中一类有免疫原性的抗原呈递成纤维细胞
NCB|心咽发育多样化的单细胞转录轨迹分析
七龙珠|召唤一份单细胞数据库汇总
用了这么多年的PCA可视化竟然是错的!!!
单细胞预测Doublets软件包汇总-过渡态细胞是真的吗?
Seurat亮点之细胞周期评分和回归
cellassign:用于肿瘤微环境分析的单细胞注释工具(9月Nature)
Nature重磅综述 |关于RNA-seq,你想知道的都在这
转录组研究
39个转录组分析工具,120种组合评估(转录组分析工具哪家强-导读版)
39个转录组分析工具,120种组合评估(转录组分析工具大比拼 (完整翻译版))
无参转录组分析工具评估和流程展示
120分的转录组试题(第一份答案)
120分的转录组试题(第二份答案)
120分的转录组试题(第三份答案)
DESeq2差异基因分析和批次效应移除
美女教授带你从统计学视角看转录组分析
整合QC质控结果的利器——MultiQC
自从用了这个神器,大规模RNA-seq数据挖掘我也可以
NGS基础 - FASTQ格式解释和质量评估
NGS基础 - 高通量测序原理
NGS基础 - 参考基因组和基因注释文件
NGS基础 - GTF/GFF文件格式解读和转换
NGS基础 - 测序原始数据下载
Illumina测序仪比较和各种测序应用模式图,助力了解高通量测序
生信分析过程中这些常见文件的格式以及查看方式你都知道吗?
本地安装UCSC基因组浏览器
测序数据可视化 (一)
IGV基因组浏览器可视化高通量测序数据
高通量数据分析必备-基因组浏览器使用介绍 - 1
高通量数据分析必备-基因组浏览器使用介绍 - 2
高通量数据分析必备-基因组浏览器使用介绍 - 3
测序文章数据上传找哪里
GO、GSEA富集分析一网打进
GSEA富集分析 - 界面操作
无需写代码的高颜值富集分析神器
去东方,最好用的在线GO富集分析工具
没钱买KEGG怎么办?REACTOME开源通路更强大
超简便的国产lncRNA预测工具LGC
我想做信号通路分析,但我就是不想学编程
一个逆天的small RNA-seq数据挖掘神器
一文掌握GSEA,超详细教程
这个只需一步就可做富集分析的网站还未发表就被CNS等引用超过350次
什么,你算出的P-value看上去像齐天大圣变的庙?
单基因GSEA怎么做?
赠你一只金色的眼 - 富集分析和表达数据可视化
NCB|心咽发育多样化的单细胞转录轨迹分析
用了这么多年的PCA可视化竟然是错的!!!
如何火眼金睛鉴定那些单细胞转录组中的混杂因素
Nature重磅综述 |关于RNA-seq,你想知道的都在这
往期精品
画图三字经 生信视频 生信系列教程
心得体会 TCGA数据库 Linux Python
高通量分析 免费在线画图 测序历史 超级增强子
生信学习视频 PPT EXCEL 文章写作 ggplot2
海哥组学 可视化套路 基因组浏览器
色彩搭配 图形排版 互作网络
自学生信 2019影响因子 GSEA 单细胞
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集

