GEO芯片数据分析更新(补富集分析与WGCNA)
GEO数据挖掘,表达芯片分析
举例:王同学近期拟通过生物信息学相关软件与数据库来探讨女性非抽烟者的非小细胞肺癌预后相关的显著性基因及潜在的治疗靶点,他在NCBI上查询到了1套芯片数据GSE19804。请帮助他完成该项目的设计与分析。
上一篇博文我发现有两个问题,一个是分组问题,PCA结果不好;另一个是筛选出的差异基因太多,之前是R中下载GSE,后来我发现可以直接下载matrix和GPL注释文件,这次还是GSE19804这个数据,再重新分析下(这次新增加KEGG和KO富集分析,WGCNA分析):
***备注:***其实这里所有用到R的分析,都可以用在线分析工具如GEO2R、David、image GP、微生信等在线分析工具完成,比如这篇博文中WGCNA我就是用的在线工具,其他部分如果想了解下R代码的话,可以参考我写出的代码
一、一般流程
1、找数据,找到GSE编号
2、下载数据:包括表达矩阵、临床信息、分组信息
3、数据探索:分组之间是否有差异,PCA,热图
4、limma差异分析及可视化:P值、logFC、火山图、热图
5、富集分析KEGG、GO
二、数据读取与预处理
基本过程和上一篇博文是一致的,用到的R包:
######################软件包下载###############################
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("impute")
BiocManager::install("limma")
install.packages("ggplot2")
install.packages("ggrepel")
##############################################################
library(impute)
library(limma)
library(ggplot2)
library(ggrepel)
logFoldChange=2#阈值自己看着调
adjustP=0.05
1、数据导入
首先直接上代码:
ann <- read.table("D:/生信/GPL570-55999.txt",sep = "\t",header = T,fill = TRUE,quote = "")
data <- read.table("D:/生信/GSE19804_series_matrix.txt",sep = "\t",header = T)
***备注:***下载下来的东西可能很多,不需要全都读取,可以手动删掉一部分,从series_matrix_table_begin开始保留就行(如下图):

在读入下载好的表达矩阵时,为什么要加那么多参数才能下载成功?我们首先需要在电脑上解压并打开文本文件,根据文件的样子选择参数:

如果报错:列的数目比列的名字要多,就试试下面这段代码:
data = read.table(file="D:/生信/GSE19804_series_matrix.txt",
header = T,sep = "\t",quote = "",fill = T,
comment.char = "!")
2、基因ID转换
***理论:***基因ID之间的转换,我们下载的数据通常使用的是不同的芯片探针,它们有不同的探针ID(probe_id)我们需要把它转化成entrez ID或symbol ID才能被大众认知;
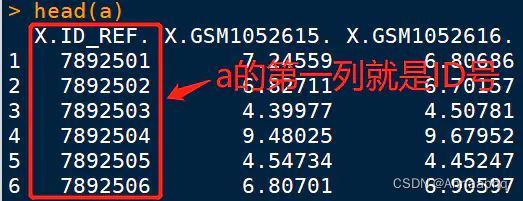
注意:并不是所有都给的是探针ID,还有很多数据给的是转录本ID,这也是我为什么说是标准流程,但是不能覆盖所有
这里有两种方法,一种是上一篇博文已经介绍的,用R获取芯片探针与基因的对应关系三部曲-bioconductor里搜索GPL6244所对应的R包;另一种就是这篇博文里提到的代码,即事先下载GPL文件,直接合并处理
2.1 GPL信息提取
直接上代码:
#目的是提取GPL文件中的3列,即ID、Gene_Symbol、Eesembl,关键是ID、Gene_Symbol一定要提取,这里提取两列
ann <- ann[,c(1,11)]
2.2 ID合并
这里有一个问题,GPL中提取出来的ID没有引号,但表达矩阵中是有引号的:


所以这里需要先去掉引号,代码为:
## nrow(AA)表示矩阵的行数
for (i in 1:nrow(data) ){
x=data[i,1] # 赋值
x=as.character(x) #化作字符串
a=gsub('["]', '', x) #去双引号
data[i,1]=a #给矩阵重新赋值
}
合并方法1:
data <- merge(ann,data,by.x = "ID",by.y = "ID_REF")
data <- data[,c(2,4:9)]
data <- as.matrix(data)
rownames(data) <- data[,1]
exp <- data[,2:ncol(data)]
dimnames <- list(rownames(exp),colnames(exp))#提取行名和列名
exp <- matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
合并方法2:(我用的是这个)
检查一下有没有对应不上的探针:
length(unique(ann$Gene.Symbol))
tail(sort(table(ann$Gene.Symbol)))
table(sort(table(ann$Gene.Symbol)))
rownames(data)= data[,1]
data = data[,-1]
table(rownames(data) %in% ann$ID)
均可以对应上,对应不上的处理方法可以看我附在文末的参考资料,里面很详细

使用match函数把ids里的探针顺序改一下,使ids里探针顺序和我们表达矩阵的顺序完全一样
ann=ann[match(rownames(data),ann$ID),]
然后进行合并:
tmp = by(data,
ann$Gene.Symbol,
function(x) rownames(x)[which.max(rowMeans(x))])
dim(data)
probes = as.character(tmp)
data = data[rownames(data) %in% probes,]
dim(data)
rownames(data)=ann[match(rownames(data),ann$ID),2]#过滤有多个探针的基因
结果如下:

提取行名与列名,并转为表达矩阵:
exp <- data[,1:ncol(data)]#和上方从2开始不一样,需注意
dimnames <- list(rownames(exp),colnames(exp))#提取行名和列名
exp <- matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
3、填充缺失值
直接上代码:
#####缺失值填充#####
mat=impute.knn(exp)
rt=mat$data
rt=avereps(rt)
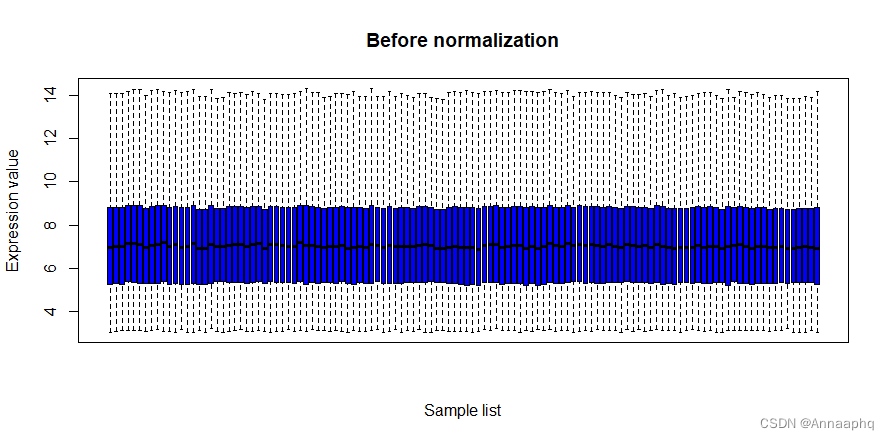
4、查看校正情况
直接上代码
#####normalize#####
#pdf(file="rawBox.pdf")
boxplot(rt,col = "blue",main = "Before normalization",
xlab = "Sample list",
ylab = "Expression value",xaxt = "n",outline = F)
#dev.off()
rt=normalizeBetweenArrays(as.matrix(rt))
#pdf(file="normalBox.pdf")
boxplot(rt,col = "red",main = "Normalization",
xlab = "Sample list",
ylab = "Expression value",xaxt = "n",outline = F)
#dev.off()
#rt=log2(rt+1)
这个芯片数据处理的比较规则,基本不需要校正:
三、差异性分析
1、火山图
首先进行分组:
GEO中搜索GSE19804,发现可以分为2组,癌组织与正常组织样本,样本量60:60

class <- c(rep("dis",60),rep("con",60)) #需要根据实验设计进行修改
design <- model.matrix(~0+factor(class))
colnames(design) <- c("con","dis")
fit <- lmFit(rt,design)
cont.matrix<-makeContrasts(dis-con,levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
allDiff=topTable(fit2,adjust='fdr',number=200000)
allDiff$gene_id <- rownames(allDiff)
allDiff <- allDiff[, colnames(allDiff)[c(7,1:6)]]
write.table(allDiff,file="D:/生信/limmaTab.xls",sep="\t",quote=F,row.names = F)
#write table(adjp)
diffSig <- allDiff[with(allDiff, (abs(logFC)>logFoldChange & adj.P.Val < adjustP )), ]
write.table(diffSig,file="D:/生信/diff_adj.xls",sep="\t",quote=F,row.names = F)
diffUp <- allDiff[with(allDiff, (logFC>logFoldChange & adj.P.Val < adjustP )), ]
write.table(diffUp,file="D:/生信/up_adj.xls",sep="\t",quote=F,row.names = F)
diffDown <- allDiff[with(allDiff, (logFC<(-logFoldChange) & adj.P.Val < adjustP )), ]
write.table(diffDown,file="D:/生信/down_adj.xls",sep="\t",quote=F,row.names = F)
hmExp=rt[rownames(diffSig),]
diffExp=rbind(id=colnames(hmExp),hmExp)
write.table(diffExp,file="D:/生信/diffExp_adj.txt",sep="\t",quote=F,col.names=F)
#write table(pvalue)
diffSig <- allDiff[with(allDiff, (abs(logFC)>logFoldChange & P.Value < adjustP )), ]
write.table(diffSig,file="D:/生信/diff_pvale.xls",sep="\t",quote=F,row.names = F)
diffUp <- allDiff[with(allDiff, (logFC>logFoldChange & P.Value < adjustP )), ]
write.table(diffUp,file="D:/生信/up_pvale.xls",sep="\t",quote=F,row.names = F)
diffDown <- allDiff[with(allDiff, (logFC<(-logFoldChange) & P.Value < adjustP )), ]
write.table(diffDown,file="D:/生信/down_pvale.xls",sep="\t",quote=F,row.names = F)
hmExp=rt[rownames(diffSig),]
diffExp=rbind(id=colnames(hmExp),hmExp)
write.table(diffExp,file="D:/生信/diffExp_pvale.txt",sep="\t",quote=F,col.names=F)
write table(pvalue)是防止根据前一个校正的的结果没有显著性,是另一种方法

2、表达矩阵分布图
# 准备画图所需数据
library(reshape2)
head(exp)
exp_L = melt(exp)
head(exp_L)
colnames(exp_L)=c('symbol','sample','value')
head(exp_L)
# 获得分组信息
class <- c(rep("dis",60),rep("con",60))
exp_L$group = rep(class,each=nrow(exp))
head(exp_L)
# ggplot2画图
library(ggplot2)
p = ggplot(exp_L,
aes(x=sample,y=value,fill=group))+geom_boxplot()
print(p)
##boxplot图精修版
p=ggplot(exp_L,aes(x=sample,y=value,fill=group))+geom_boxplot()
p=p+stat_summary(fun.y="mean",geom="point",shape=23,size=3,fill="red")
p=p+theme_set(theme_set(theme_bw(base_size=20)))
p=p+theme(text=element_text(face='bold'),axis.text.x=element_text(angle=30,hjust=1),axis.title=element_blank())
print(p)

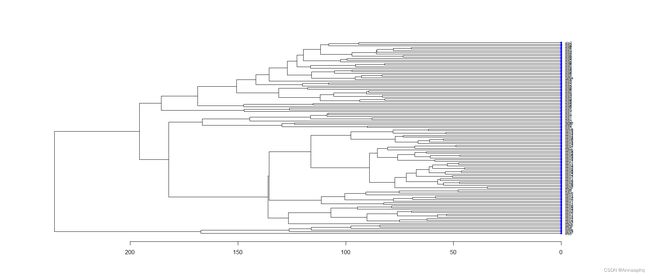
3、检查样本分组信息
检查样本分组信息,一般看PCA图,hclust图
3.1 hclust图
# 更改表达矩阵列名
head(exp)
colnames(exp) = paste(class,1:6,sep='')
head(exp)
# 定义nodePar
nodePar <- list(lab.cex = 0.6, pch = c(NA, 19),
cex = 0.7, col = "blue")
# 聚类
hc=hclust(dist(t(exp)))
par(mar=c(5,5,5,10))
# 绘图
plot(as.dendrogram(hc), nodePar = nodePar, horiz = TRUE)
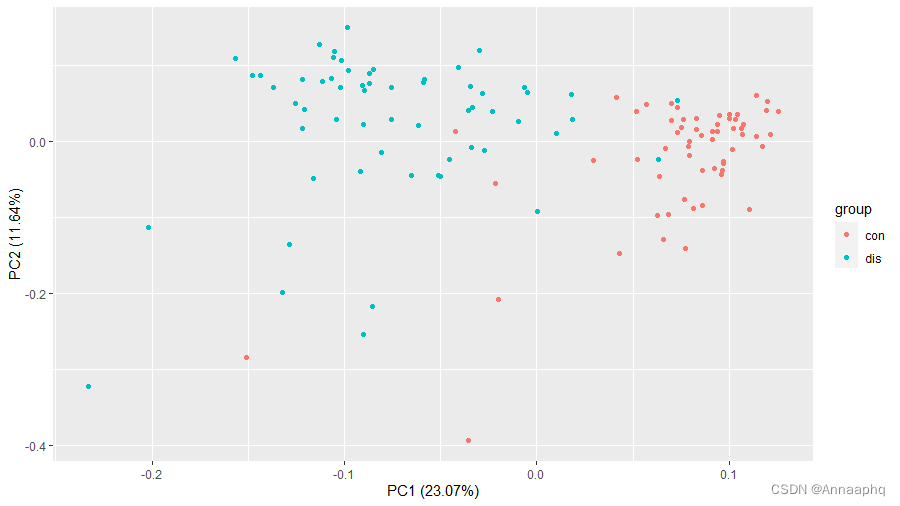
3.2 PCA
library(ggfortify)
# 互换行和列,再dim一下
df=as.data.frame(t(exp))
# 不要view df,列太多,软件会卡住;
dim(df)
dim(exp)
exp[1:6,1:6]
df[1:6,1:6]
df$group=class
autoplot(prcomp( df[,1:(ncol(df)-1)] ), data=df,colour = 'group')
和上一篇博文相比,分类情况好多了,该分开的分开了,该聚在一起的聚在一起了,数据很好,符合预期
4、画热图
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
allDiff=topTable(fit2,adjust='fdr',number=200000)
allDiff$gene_id <- rownames(allDiff)
allDiff <- allDiff[, colnames(allDiff)[c(7,1:6)]]
#截止到这里的代码都是前面画火山图出现过的
#下面为新代码
nrDEG = na.omit(allDiff)
head(nrDEG)
library(pheatmap)
choose_gene = head(rownames(nrDEG),25)
choose_matrix = exp[choose_gene,]
choose_matrix = t(scale(t(choose_matrix)))
pheatmap(choose_matrix)
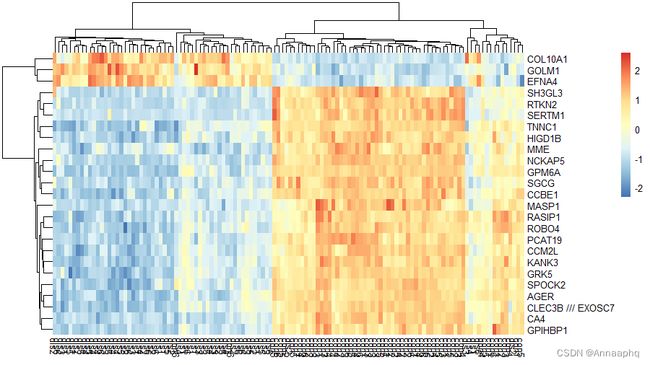
四、富集分析
1、KO富集
#####################################KO富集分析######################################
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
library(org.Hs.eg.db)
library(clusterProfiler)
library(dplyr)
f = diffSig #diffSig是火山图出找出的差异表达基因
x <-f[,1] #取所需的列进行后续分析
hg<-bitr(x,fromType="SYMBOL",
toType=c("ENTREZID","ENSEMBL","SYMBOL"),
OrgDb="org.Hs.eg.db") #用bitr函数进行ID转换,使用bioconductor系列包进行
head(hg) #查看hg信息,前3列包括SYMBOL、ENTREZID、ENSEMBL
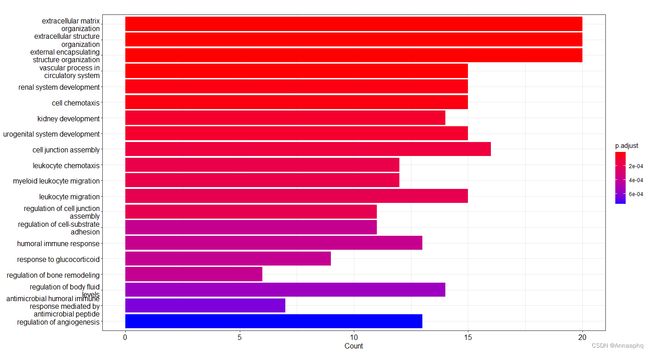
go <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='ALL',
pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
keyType = 'ENTREZID') #进行GO富集,求得P值和Q值,并用BH方法对值进行调整
dim(go) #查看富集结果
write.csv(go,file="D:/生信/go.csv") #导出富集结果
barplot(go,showCategory=20,drop=T) #柱状图
dotplot(go,showCategory=20) #气泡图
emapplot(go) #网络图
cnetplot(go, showCategory = 5) #基因与GOTerm网络关系图


2、KEGG富集
#####################################KEGG富集分析######################################
goCC <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='CC',pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
keyType = 'ENTREZID') #对CC进行富集
goBP <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='BP',pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,keyType = 'ENTREZID') #对BP进行富集
goMF <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='MF',pAdjustMethod = 'BH',pvalueCutoff = 0.05,
qvalueCutoff = 0.2,keyType = 'ENTREZID') #对MF进行富集
barplot(go, split="ONTOLOGY")+ facet_grid(ONTOLOGY~.,scale="free")
kegg <- enrichKEGG(hg$ENTREZID,
organism = 'hsa',
keyType = 'kegg',
pvalueCutoff = 0.05,
pAdjustMethod = 'BH',
minGSSize = 3,
maxGSSize = 500,
qvalueCutoff = 0.2,
use_internal_data = FALSE) #对KEGG进行富集
write.csv(kegg,file = "D:/生信/kegg.csv") #导出富集结果
dim(kegg) #查看富集结果
dotplot(kegg, showCategory=20) #气泡图
barplot(kegg,showCategory=20,drop=T) #柱状图
browseKEGG(kegg, "hsa03728") #pathway中标记的基因会链接到官网



五、WGCNA加共表达网络分析
这里写出基因表达矩阵,用在线工具imageGP做的:
写出代码:
write.table(exp,file="D:/生信/exp.xls",sep="\t",quote=F,row.names = T)

六、所有代码汇总
######################软件包下载###############################
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("impute")
BiocManager::install("limma")
install.packages("ggplot2")
install.packages("ggrepel")
##############################################################
library(impute)
library(limma)
library(ggplot2)
library(ggrepel)
logFoldChange=2
adjustP=0.05
#####数据导入#####
ann <- read.table("D:/生信/GPL570-55999.txt",sep = "\t",header = T,fill = TRUE,quote = "")
data <- read.table("D:/生信/GSE19804_series_matrix.txt",sep = "\t",header = T)#这一行报错可以用:
data = read.table(file="D:/生信/GSE19804_series_matrix.txt",
header = T,sep = "\t",quote = "",fill = T,
comment.char = "!")
#####ID提取+去双引号#####
ann <- ann[,c(1,11)]
for (i in 1:nrow(data) ){
x=data[i,1] # 赋值
x=as.character(x) #化作字符串
a=gsub('["]', '', x) #去双引号
data[i,1]=a #给矩阵重新赋值
}
#####ID合并方法1(是我看到别人做的,自己做的话需要看看参数是否需要调整)#####
data <- merge(ann,data,by.x = "ID",by.y = "ID_REF")
data <- data[,c(2,4:9)]
data <- as.matrix(data)
rownames(data) <- data[,1]
exp <- data[,2:ncol(data)]
dimnames <- list(rownames(exp),colnames(exp))#提取行名和列名
exp <- matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
#####ID合并方法2#####
length(unique(ann$Gene.Symbol))
tail(sort(table(ann$Gene.Symbol)))
table(sort(table(ann$Gene.Symbol)))
rownames(data)= data[,1]
data = data[,-1]
table(rownames(data) %in% ann$ID)#检查有无对应不上的探针
ann=ann[match(rownames(data),ann$ID),]
tmp = by(data,
ann$Gene.Symbol,
function(x) rownames(x)[which.max(rowMeans(x))])
probes = as.character(tmp)
dim(data)
data = data[rownames(data) %in% probes,]
dim(data)
rownames(data)=ann[match(rownames(data),ann$ID),2]
exp <- data[,1:ncol(data)]
dimnames <- list(rownames(exp),colnames(exp))#提取行名和列名
exp <- matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
#####缺失值填充#####
mat=impute.knn(exp)
rt=mat$data
rt=avereps(rt)
#####normalize#####
#pdf(file="rawBox.pdf")
boxplot(rt,col = "blue",main = "Before normalization",
xlab = "Sample list",
ylab = "Expression value",xaxt = "n",outline = F)
#dev.off()
rt=normalizeBetweenArrays(as.matrix(rt))
#pdf(file="normalBox.pdf")
boxplot(rt,col = "red",main = "Normalization",
xlab = "Sample list",
ylab = "Expression value",xaxt = "n",outline = F)
#dev.off()
#rt=log2(rt+1)
##########################差异分析##########################
class <- c(rep("dis",60),rep("con",60)) #需要根据实验设计进行修改
design <- model.matrix(~0+factor(class))
colnames(design) <- c("con","dis")
fit <- lmFit(rt,design)
cont.matrix<-makeContrasts(dis-con,levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
allDiff=topTable(fit2,adjust='fdr',number=200000)
allDiff$gene_id <- rownames(allDiff)
allDiff <- allDiff[, colnames(allDiff)[c(7,1:6)]]
write.table(allDiff,file="D:/生信/limmaTab.xls",sep="\t",quote=F,row.names = F)
#write table(adjp)
diffSig <- allDiff[with(allDiff, (abs(logFC)>logFoldChange & adj.P.Val < adjustP )), ]
write.table(diffSig,file="D:/生信/diff_adj.xls",sep="\t",quote=F,row.names = F)
diffUp <- allDiff[with(allDiff, (logFC>logFoldChange & adj.P.Val < adjustP )), ]
write.table(diffUp,file="D:/生信/up_adj.xls",sep="\t",quote=F,row.names = F)
diffDown <- allDiff[with(allDiff, (logFC<(-logFoldChange) & adj.P.Val < adjustP )), ]
write.table(diffDown,file="D:/生信/down_adj.xls",sep="\t",quote=F,row.names = F)
hmExp=rt[rownames(diffSig),]
diffExp=rbind(id=colnames(hmExp),hmExp)
write.table(diffExp,file="D:/生信/diffExp_adj.txt",sep="\t",quote=F,col.names=F)
#write table(pvalue)
diffSig <- allDiff[with(allDiff, (abs(logFC)>logFoldChange & P.Value < adjustP )), ]
write.table(diffSig,file="D:/生信/diff_pvale.xls",sep="\t",quote=F,row.names = F)
diffUp <- allDiff[with(allDiff, (logFC>logFoldChange & P.Value < adjustP )), ]
write.table(diffUp,file="D:/生信/up_pvale.xls",sep="\t",quote=F,row.names = F)
diffDown <- allDiff[with(allDiff, (logFC<(-logFoldChange) & P.Value < adjustP )), ]
write.table(diffDown,file="D:/生信/down_pvale.xls",sep="\t",quote=F,row.names = F)
hmExp=rt[rownames(diffSig),]
diffExp=rbind(id=colnames(hmExp),hmExp)
write.table(diffExp,file="D:/生信/diffExp_pvale.txt",sep="\t",quote=F,col.names=F)
##########################绘制火山图##########################
#绘制火山图(adjp筛选)
allDiff[is.na(allDiff)] <- 0
allDiff$change = ifelse(allDiff$adj.P.Val < adjustP & abs(allDiff$logFC) >= logFoldChange,
ifelse(allDiff$logFC> logFoldChange ,'Up','Down'),
'Stable')
pdf("volcanol_FDR.pdf")
ggplot(allDiff,
aes(x = logFC,
y = -log10(adj.P.Val),
colour=change)) +
geom_point(alpha=0.4, size=1) +
scale_color_manual(values=c("#546de5", "#d2dae2","#ff4757"))+
geom_vline(xintercept=c(-1,1),lty=4,col="black",lwd=0.8) +
geom_hline(yintercept = -log10(adjustP),lty=4,col="black",lwd=0.8) +
labs(x="log2(fold change)",
y="-log10 (FDR)")+
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
legend.position="right",
legend.title = element_blank()
)
dev.off()
#绘制火山图(pvalue筛选)
allDiff[is.na(allDiff)] <- 0
allDiff$change = ifelse(allDiff$P.Value < adjustP & abs(allDiff$logFC) >= logFoldChange,
ifelse(allDiff$logFC> logFoldChange ,'Up','Down'),
'Stable')
pdf("volcanol_pvalue.pdf")
ggplot(allDiff,
aes(x = logFC,
y = -log10(P.Value),
colour=change)) +
geom_point(alpha=0.4, size=1) +
scale_color_manual(values=c("#546de5", "#d2dae2","#ff4757"))+
geom_vline(xintercept=c(-1,1),lty=4,col="black",lwd=0.8) +
geom_hline(yintercept = -log10(adjustP),lty=4,col="black",lwd=0.8) +
labs(x="log2(fold change)",
y="-log10 (pvalue)")+
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
legend.position="right",
legend.title = element_blank()
)
dev.off()
##绘制带有基因名称的火山图
allDiff[is.na(allDiff)] <- 0
allDiff$change = ifelse(allDiff$P.Value < adjustP & abs(allDiff$logFC) >= logFoldChange,
ifelse(allDiff$logFC> logFoldChange ,'Up','Down'),
'Stable')
allDiff$label = ifelse(allDiff$P.Value < adjustP & abs(allDiff$logFC) >= 2.5, as.character(allDiff$gene_id),"")
pdf("volcanol_gene.pdf")
ggplot(allDiff,
aes(x = logFC,
y = -log10(P.Value),
colour=change)) +
geom_point(alpha=0.4, size=1) +
scale_color_manual(values=c("#546de5", "#d2dae2","#ff4757"))+
geom_vline(xintercept=c(-1,1),lty=4,col="black",lwd=0.8) +
geom_hline(yintercept = -log10(adjustP),lty=4,col="black",lwd=0.8) +
labs(x="log2(fold change)",
y="-log10 (FDR)")+
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
legend.position="right",
legend.title = element_blank()
)+
geom_text_repel(data = allDiff, aes(x = logFC,
y = -log10(P.Value),
label = label),
size = 3,box.padding = unit(0.8, "lines"),
point.padding = unit(0.8, "lines"),
show.legend = FALSE)
dev.off()
########################表达矩阵分布图######################
# 准备画图所需数据
library(reshape2)
head(exp)
exp_L = melt(exp)
head(exp_L)
colnames(exp_L)=c('symbol','sample','value')
head(exp_L)
# 获得分组信息
class <- c(rep("dis",60),rep("con",60))
exp_L$group = rep(class,each=nrow(exp))
head(exp_L)
# ggplot2画图
library(ggplot2)
p = ggplot(exp_L,
aes(x=sample,y=value,fill=group))+geom_boxplot()
print(p)
##boxplot图精修版
p=ggplot(exp_L,aes(x=sample,y=value,fill=group))+geom_boxplot()
p=p+stat_summary(fun.y="mean",geom="point",shape=23,size=3,fill="red")
p=p+theme_set(theme_set(theme_bw(base_size=20)))
p=p+theme(text=element_text(face='bold'),axis.text.x=element_text(angle=30,hjust=1),axis.title=element_blank())
print(p)
##########################检查样本分组信息##################
#hclust#
# 更改表达矩阵列名
head(exp)
colnames(exp) = paste(class,1:6,sep='')
head(exp)
# 定义nodePar
nodePar <- list(lab.cex = 0.6, pch = c(NA, 19),
cex = 0.7, col = "blue")
# 聚类
hc=hclust(dist(t(exp)))
par(mar=c(5,5,5,10))
# 绘图
plot(as.dendrogram(hc), nodePar = nodePar, horiz = TRUE)
#PCA
library(ggfortify)
# 互换行和列,再dim一下
df=as.data.frame(t(exp))
# 不要view df,列太多,软件会卡住;
dim(df)
dim(exp)
exp[1:6,1:6]
df[1:6,1:6]
df$group=class
autoplot(prcomp( df[,1:(ncol(df)-1)] ), data=df,colour = 'group')
#####################热图#########################
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
allDiff=topTable(fit2,adjust='fdr',number=200000)
allDiff$gene_id <- rownames(allDiff)
allDiff <- allDiff[, colnames(allDiff)[c(7,1:6)]]
#截止到这里的代码都是前面画火山图出现过的
#下面为新代码
nrDEG = na.omit(allDiff)
head(nrDEG)
library(pheatmap)
choose_gene = head(rownames(nrDEG),25)
choose_matrix = exp[choose_gene,]
choose_matrix = t(scale(t(choose_matrix)))
pheatmap(choose_matrix)
#####################################KO富集分析######################################
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
library(org.Hs.eg.db)
library(clusterProfiler)
library(dplyr)
f = diffSig #diffSig是火山图出找出的差异表达基因
x <-f[,1] #取所需的列进行后续分析
hg<-bitr(x,fromType="SYMBOL",
toType=c("ENTREZID","ENSEMBL","SYMBOL"),
OrgDb="org.Hs.eg.db") #用bitr函数进行ID转换,使用bioconductor系列包进行
head(hg) #查看hg信息,前3列包括SYMBOL、ENTREZID、ENSEMBL
go <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='ALL',
pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
keyType = 'ENTREZID') #进行GO富集,求得P值和Q值,并用BH方法对值进行调整
dim(go) #查看富集结果
write.csv(go,file="D:/生信/go.csv") #导出富集结果
barplot(go,showCategory=20,drop=T) #柱状图
dotplot(go,showCategory=20) #气泡图
emapplot(go) #网络图
cnetplot(go, showCategory = 5) #基因与GOTerm网络关系图
#####################################KEGG富集分析######################################
goCC <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='CC',pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
keyType = 'ENTREZID') #对CC进行富集
goBP <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='BP',pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,keyType = 'ENTREZID') #对BP进行富集
goMF <- enrichGO(hg$ENTREZID,OrgDb = org.Hs.eg.db,
ont='MF',pAdjustMethod = 'BH',pvalueCutoff = 0.05,
qvalueCutoff = 0.2,keyType = 'ENTREZID') #对MF进行富集
barplot(go, split="ONTOLOGY")+ facet_grid(ONTOLOGY~.,scale="free")
kegg <- enrichKEGG(hg$ENTREZID,
organism = 'hsa',
keyType = 'kegg',
pvalueCutoff = 0.05,
pAdjustMethod = 'BH',
minGSSize = 3,
maxGSSize = 500,
qvalueCutoff = 0.2,
use_internal_data = FALSE) #对KEGG进行富集
write.csv(kegg,file = "D:/生信/kegg.csv") #导出富集结果
dim(kegg) #查看富集结果
dotplot(kegg, showCategory=20) #气泡图
barplot(kegg,showCategory=20,drop=T) #柱状图
browseKEGG(kegg, "hsa03728") #pathway中标记的基因会链接到官网
资料来源:
https://zhuanlan.zhihu.com/p/344426350
https://mp.weixin.qq.com/s/_izW1rqzU2y229CaZSHw8g
http://www.ehbio.com/Cloud_Platform/front/#/
备注:另一篇博文“单组率得meta分析”中参考资料来源正文中忘记加了,这里补一下:
主要是3个公众号:医咖会、逍遥君自习室、尔云间meta分析,链接如下:
https://mp.weixin.qq.com/s/uZmHCZBReRFiiI1P5oSzRg
https://mp.weixin.qq.com/s/xC4l46b_8jGj-FAs35VhUg
https://mp.weixin.qq.com/s/Ou99cA3Y1t68zNx7PcpeIA