论文笔记:EGAT: Edge Aggregated Graph Attention Networks and Transfer Learning
文章目录
- 论文概况
- 摘要
- 1 介绍
- 2 方法
-
- 2.1 特征表示
-
- 2.1.1 蛋白质的图表示
- 2.1.3 边特征表示
- 2.2 EGAT的结构
-
- 2.2.2 边缘聚合图关注层
- 2.2.3 预测概率
- 2.2.4 EGAT结构图
- 2.3 EGAT端到端的结构
- 2.4 GAT-PPI:基于GAT的没有边聚合PPI预测
- 3 结果和讨论
-
- 3.1 数据
- 3.2 结果
-
- 3.2.1 与其他先进方法独立测试比较
- 3.3 长期相互作用对PPI站点预测的影响
-
- 3.4 基于 ProtBERT特征的迁移学习的影响
- 3.5 边的可解释性和注意力得分
- 4 总结
- 5 启发
论文概况
原文链接:https://www.biorxiv.org/content/10.1101/2020.11.07.372466v2.abstract
原文代码链接: https://github.com/Sazan-Mahbub/EGAT
摘要
边缘聚集图注意网络(EGAT:Edge Aggregated Graph Attention Networks)一种高精度的基于深度学习蛋白质相互作用位点的预测方法,引入了一个新的边缘聚合图注意网络来有效地利用结构信息。首次将迁移学习应用于PPI站点预测。提出的边缘聚合网络,以及迁移学习,已经取得了显著的改进比最佳替代方法。此外EGAT提供了一个比典型的黑盒深度神经网络更易于解释的框架。
1 介绍
氨基酸残基相互作用的定义:绝对溶剂可及性小于1 A ˚ 2 Å^2 A˚2
Graph neural networks (GNN)编码结构特征:已经应用于成对结合位点的预测,但是还没有应用于单个的氨基酸残基。
Graph Attention Network (GAT)编码结构特征:采用了注意力机制,但是仅仅只考虑了节点,没有考虑边的特征,GAT缺乏利用边缘特征中可能编码的丰富结构信息的能力。
边缘聚集图注意网络(EGAT:Edge Aggregated Graph Attention Networks):有效利用编码在边缘特征中的结构信息。在预训练的模型中成功用迁移学习,EGAT还有助于构建可解释的深度学习模型。
2 方法
2.1 特征表示
2.1.1 蛋白质的图表示
EGAT模型是一个基于图神经网络的体系结构,将数据集中每个蛋白质P的三维结构表示为有向k近邻图G。
- V (G) :表示氨基酸残基节点
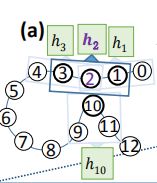
- N i N_i Ni:表示节点i的领域,i一共有K个领域节点,如下图K=3。任何两个节点(残基)之间的距离是通过它们的平均原子之间的距离来计算的(使用PDB文件中的原子坐标。
输入的序列 X = X 1 , X 2 , . . . . X N {X=X_1,X_2,....X_N} X=X1,X2,....XN通过ProtBERT进行编码产生节点特征向量{ q = q 1 , q 2 . . . q N q=q_1,q_2...q_N q=q1,q2...qN}, q i ∈ d p r o t b e r t q_i∈d_{protbert} qi∈dprotbert( d p r o t b e r t d_{protbert} dprotbert=1024),与其他方法相比,ProtBERT在残基水平分类任务上取得了更好的性能,EGAT与ProtTrans中提供的预训练语言模型无关,这意味着ProtTrans中的其他合适的语言模型也可以在我们的模型中使用。
2.1.3 边特征表示
在蛋白质P的有向图用G表示,使用边的两个特征:
1.残基i和j之间的距离 D i j D_{ij} Dij,它是通过取它们原子之间的平均距离来计算的
2. 残基i和j的相对取向 θ i j θ_{ij} θij,计算为这两个残基通过α碳原子( C α C_α Cα)的平面的表面法线之间夹角的绝对值.
相关的参数;
E i j E_{ij} Eij:蛋白质的边
氨基酸残基原子之间的平均距离来计算理解

PDB文件数据理解:
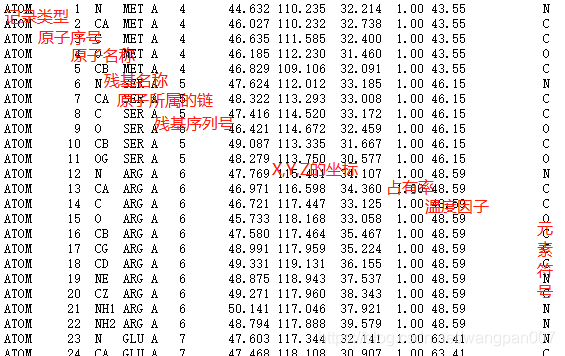
主要氨基酸缩写及短写:
蛋氨酸(Methionine): MET(M)
丝氨酸(serine):SER(S)
精氨酸(Arginine):ARG(G)
谷氨酸(Glutamic acid):GLU(E)
2.2 EGAT的结构
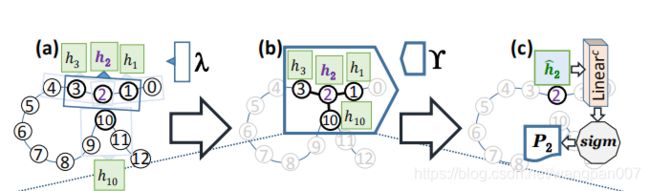
2.2.2 边缘聚合图关注层
现在我们描述最初的图关注层[44]和我们通过引入边聚合而提出的修改。特征表示h(由局部特征提取器λ产生)使用我们提出的边缘聚合图注意层Υ进行变换,以编码蛋白质的三维结构信息。各图神经网络基础架构,有一个节点的聚合过程中我所有相邻节点的特性表征聚合生成一个固定大小的新 U i U_i Ui表示为节点,然后用于进一步的计算。一个常用的聚合过程是对相邻节点的特征进行加权平均。在这个过程中,第i个节点 U i ∈ R f n U_i∈R^{f_n} Ui∈Rfn的表示由下式生成。
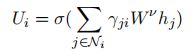
![]()
![]()
边特征 ξ i j ξ_{ij} ξij的注意力分数对的softmax规范化处理:
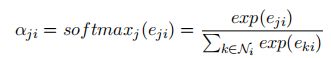
- W η W_η Wη:学习参数
- h i ˆ h^ˆ_i hiˆ :通过边缘聚合图关注层Υ,最终的特征表示。
- σ(.):激活函数
- W ε W^ε Wε:边特征通过线性层后得到的学习参数
2.2.3 预测概率
![]()
W c W^c Wc: h i ˆ h^ˆ_i hiˆ通过线性层之后学习的参数
sigm(.):sigmoid激活函数
P ˆ i Pˆ_i Pˆi:残基i与其他蛋白质相互作用的数值倾向
2.2.4 EGAT结构图
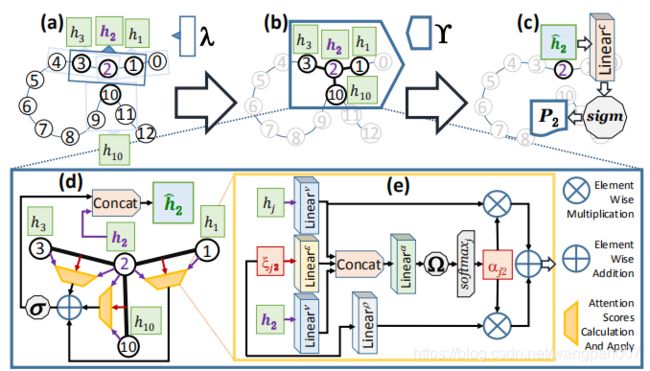
图七、 EGAT应用于含有13个残基的假蛋白的整个管道示意图(i = 0, 1, 2, . . . , 12).(a)局部特征提取器λ(窗口大小 W l o c a l W_{local} Wlocal)。(b)将边聚集图关注层Υ应用于以残基2为中心节点的邻域,残基{1,3,10}∈ N 2 N_2 N2为残基2的领域节点。©应用于节点2的最终表示 h i ˆ h^ˆ_i hiˆ的节点级分类器(由Υ生成)。这里,Sigm表示生成概率 P 2 P_2 P2的Sigmoid激活函数,通过概率 P 2 P_2 P2来判定节点2是否属于相互作用位点。(d)边缘聚合图注意层Υ的细节以展开,黄色梯形表示计算注意力分数并将其应用于节点和边的特征的模块。(e)黄色梯形的注意力机制,公式四的过程。 L i n e a r x Linear^x Linearx表示一个线性层(全连接层),学习参数 W x W^x Wx
2.3 EGAT端到端的结构
EGAT应用于具有13个残基的虚拟蛋白质输入蛋白质序列直接得到输出的结构(没有类似于降维,特征重要性等操作),并且为了该图的可读性和清晰度,显示了仅针对残基2的计算。EGAT首先将蛋白质表示为图G,其中每个节点i∈V(G)都连接到|Ni|=k个最近的具有有向边的节点。出于可读性的考虑,我们在本例中使用了 N i N_i Ni=k=3。这里 N 2 N_2 N2={1,3,10}。并不是说残基10不是顺序地更接近节点2,而是在三维空间中非常接近节点2。
EGAT具体步骤:
- 使用ProtBERT对氨基酸残基{ X = X 1 , X 2 , . . . . X N X={X_1,X_2,....X_N} X=X1,X2,....XN}进行特征编码转为特征向量{q= q 1 q_1 q1,q_2…q_N}$指定为V(G)中节点的初始特征。
- 从蛋白质的结构数据(可在pdb文件[42]中获得)计算边 E i j E_{ij} Eij的特征向量 ξ i j ξ_{ij} ξij,每个边特征向量 ξ i j ξ_{ij} ξij表示从节点j到节点i的有向边。
- 一维卷积构成的局部特征提取器λ被应用于蛋白质节点的特征表示(Q)。q通过λ生成一个低维表示{ h = h 1 , h 2 , h 3 , . . h N h ={h_1, h_2, h_3, . .h_N} h=h1,h2,h3,..hN}, h i ∈ R f n h_i ∈R^{fn} hi∈Rfn , f n < d p r o t b e r t {fn}
- 在每个邻域上应用边聚合图关注层 γ \gamma γ, γ \gamma γ生成邻域中心结点i的最终要素制图表达 h i ˆ h^ˆ_i hiˆ
- 节点级分类器为我们提供了残基2的概率值 P 2 P_2 P2,它是残基2作为接口残基(或相互作用位点)的预测倾向。
2.4 GAT-PPI:基于GAT的没有边聚合PPI预测
GAT-PPI没有任何边聚集,以显示基于图的方法相对于其他竞争方法的优越性。我们称此方法为GAT-PPI。我们使用了Deep Graph Library提供的原始实现。
3 结果和讨论
3.1 数据
- Dset_186
- Dset_164
- Dset_72
这三个数据集都是用pdb-database[42]中的蛋白质建立的,序列同源性小于25%,分辨率小于3.0˚A(用X射线晶体学解决)。由于这些数据集来自不同的研究小组,[15]将这三个数据集整合成一个融合的数据集,以确保训练集和测试集来自相同的分布。70个(16.6%)随机选择的蛋白质序列的测试集和(约83.4%)蛋白质序列用作训练集,训练集比DeePPISP多了两条蛋白质序列。
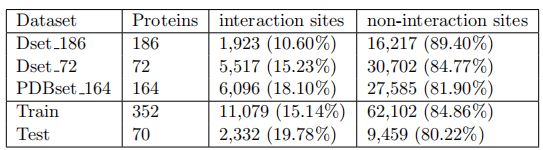
3.2 结果
3.2.1 与其他先进方法独立测试比较
作者提出的两个方法( EGAT和GAT-PPI)与其他方法进行比较
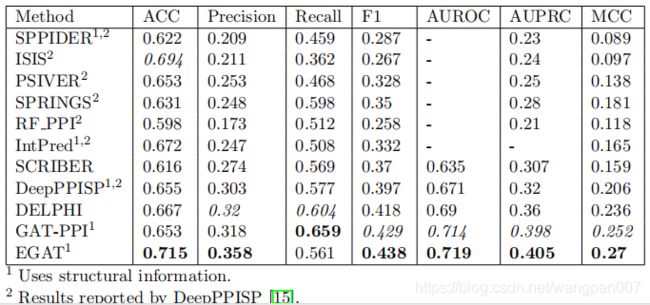
为了直观地显示预测的界面位置,图五代表蛋白(PDB ID 30UR,Chain-B)上真实和预测的(EGAT和Delphi)相互作用位点。该蛋白全长150个残基,有39个相互作用位点。它有412对非本地接触对,表明存在高水平的远程互动。
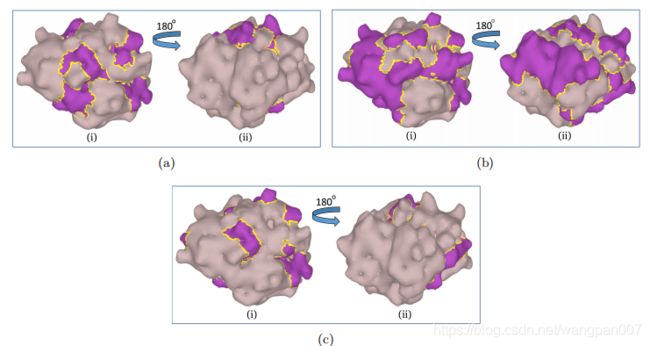
图九、(a)EGAT预测的相互作用点,(b)Delphi预测的交互站点,以及©从数据集中获得的真实相互作用位点。蛋白质表面的相互作用位点以紫色表示。每个图(a、b和c)中的左和右图像(I和II)显示两个相对的侧面(即180旋转视图)。
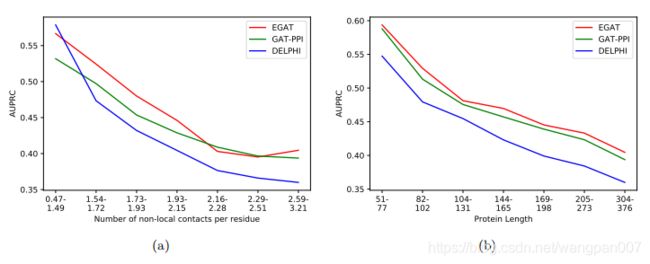
3.3 长期相互作用对PPI站点预测的影响
与DeepPPISP不同,EGAT的设计目的是为不同的残基生成不同的合适的全局特征。研究了EGAT在不同水平的长期交互作用下的表现,并与Delphi和GAT-PPI进行了比较。计算了测试集中70个蛋白质中每个残基的非本地相互作用的数量,并按升序对它们进行了排序。序列位置i和j上的两个残基如果相距至少20个残基(|i-j|≥20),则认为它们具有非局部相互作用,但根据C˚原子之间的原子距离小于8Å

把测试集它们放入七个大小相等的箱子b1,b2…b7(每个包含10个蛋白质,其中b1包含非局部相互作用水平最低的蛋白质(每个残基0.41-1.49个非局部接触),b7代表具有最高非局部相互作用水平(每个残基2.59-3.21个非局部接触)的模型条件),正如预期的那样,EGAT和Delphi的性能都随着非本地接触数量的增加而下降。然而,EGAT和Delphi之间的预测性能差异随着非本地交互级别的增加而显著增加(少数例外)。这些结果清楚地表明,通过合适的全局特征来处理非局部相互作用是改进的关键因素之一
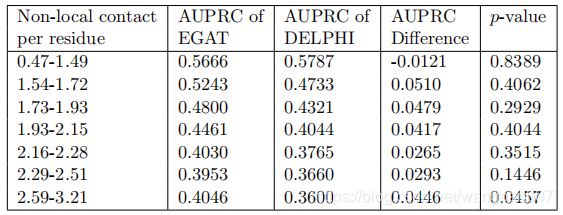
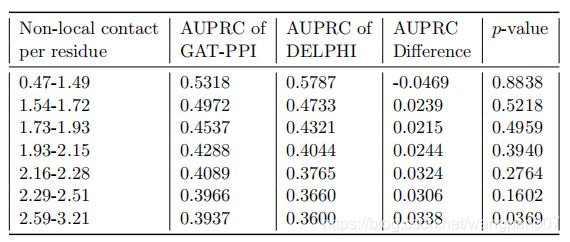
3.4 基于 ProtBERT特征的迁移学习的影响
EGAT在这两个功能集上的表现始终优于GAT-这表明无论选择哪种功能集,边缘聚合都会带来积极的影响。此外,即使没有ProtTrans特性(即,使用DeepPPISP特性集),EGAT也比DeepPPISP和DELPHI更好或一样好。
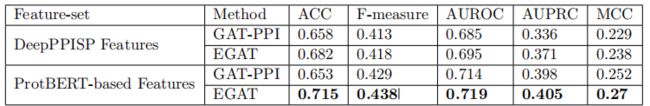
3.5 边的可解释性和注意力得分
为了评估边及其特征的可解释性,提出的基于图的模型中,研究了与有向边的源节点和目的节点相对应的数值倾向之间的相关性。具体来说,对于一条边{Eij |Eij∈E(Gprot)和i, j∈V (Gprot)},我们计算了两个有序链表之间的相关系数 P i P_i Pi和 P j P_j Pj。我们使用Scikit-Learn库中实现的pearson相关函数对整个测试集进行了这一分析。对于一个相互作用位点,其注意力得分相对较高的邻居比其注意力得分相对较低的邻居更有可能成为一个相互作用位点。
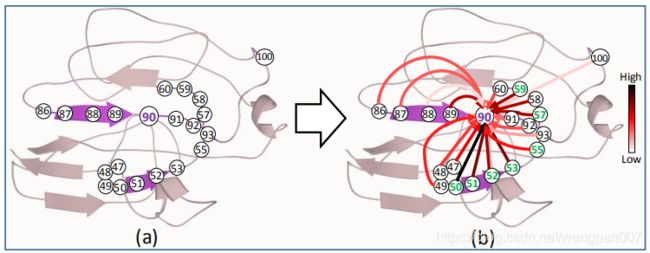
预测为相互作用位点(第50、51、52、53、55、57、59位残基)。在这些残基中,50、52、53和59是真正的相互作用位点(这四个残基与前五个注意力得分中的四个相关)。此外,具有初级序列中更接近90的非交互源节点的边(例如86、87、88、89、91、92、93)的注意力分数低于与远程交互相关联的边的注意力分数(例如50、51、52、53、59)。
仅由注意力分数建议的交互站点可能包含假阳性(例如,剩余51)和假阴性。较高的注意力分数并不一定保证一个互动网站,也不能确定另一个互动网站附近的所有互动网站都会有相对较高的注意力分数。事实上,EGAT的预测并不完全取决于注意力得分,因为它正确地预测残基58和89是非互动位点,尽管它们的相关边缘有很高的注意力得分。后续研究需要进一步研究这种基于图形的模型的可解释性,并设计一种注意力机制,使注意力得分与真实的互动地点预测更紧密地联系在一起。
4 总结
- 使用边缘聚合来扩展GAT,并证明了它在提高PPISP性能方面的有效性。
- 首次在PPISP中利用ProtBERT生成的特征集进行迁移学习。我们的实验结果表明,GAT(带或不带边聚合)明显优于其他竞争方法
- 研究了不同方法在不同模型条件下的性能,以及不同水平的长程相互作用和蛋白质长度
- 演示了EGAT模型如何有助于提高基于深度神经网络的方法的可解释性。另一个潜在的研究方向EGAT和GAT-PPI在成对PPI位点预测中的效果
5 启发
1.采用ProtBERT得到特征用传统的方法来进行学习
2.残基的不同方法三维可视化图可以学习应用
3.通过一维卷积滑动窗口的方法提取领域的特征可以应用
4.展现展现实验结果的时候如果更加准备的表达我们实验的方法好于其他先进方法