MetaGEM——构建基于基因组的代谢模型GEM
MetaGEM——构建基于基因组的代谢模型GEM
- 基础知识
-
- 代谢模型
- 基因组规模的网络代谢模型
- GEM构建工具
- MetaGEM实战
-
- 简介
- 安装
- 使用
基础知识
代谢模型
代谢分析指对生物体或体内某一特定组织所包含的所有代谢物的定量分析,并研究该代谢物组在外界干预或病理生理条件下的动态变化规律。代谢网络的重建起着至关重要的作用,该网络代表了生物与生物和非生物环境的相互作用以及将营养物质转化为生物量的能力。代谢模型已被应用于识别不同生物体之间的代谢相互作用,研究宿主与微生物组之间的相互作用,预测对抗微生物病原体的新药物靶点。
基因组规模的网络代谢模型
基因组规模的网络代谢模型(Genome-scale metabolic model,GEM),包含了某种特定生物或者是细胞基因组范围代谢反应,及其酶及基因关联的数学模型。全基因组规模的代谢网络模型:考虑目标菌株全部的基因的表达、转录、翻译过程,充分模拟生长过程的代谢变化。
基因组规模代谢网络模型构建过程是建立基因-蛋白质-生化反应之间联系的过程,涉及到基因组、蛋白质和酶、中间代谢产物、生化反应、代谢途径等诸多方面的数据。
DNA测序技术的最新进展导致数据库中可用的基因组和宏基因组序列大量增加,这进一步扩大了基因组尺度代谢网络重建的适用性。
目前有许多研究构建物种及在特定环境下的代谢网络模型,一般情况下不需要从头开始重建一个代谢网络模型,可以参考已有的代谢模型。常见物种的全基因组代谢模型(genome-scale metabolic model)可以在BiGG Models上找到,以及ModelSEED、KBase等。以及目前许多已经发表的文献中提供可下载GEM的网址。(下载.xml文件)
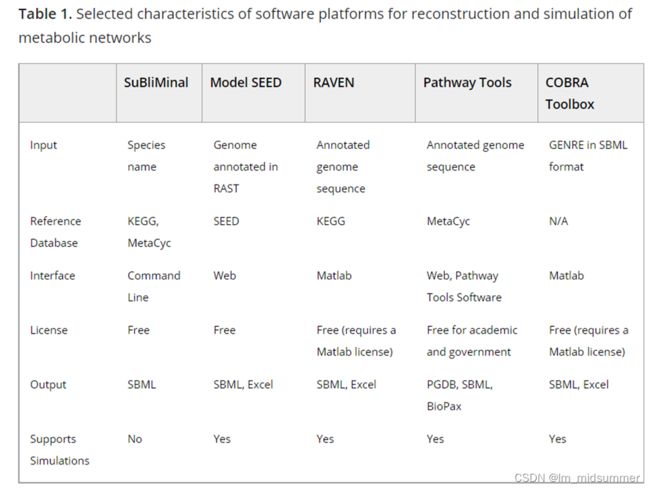
GEM构建工具
基因组计算注释的进展和在线数据库中存储的生化知识的大量增加促使开发了几种软件方法来自动化重建过程。Mendoza等人最近的一项研究综合比较了目前七种基因组级代谢重建工具(Mendoza S N, Olivier B G, Molenaar D and Teusink B 2019 A systematic assessment of current genome-scale metabolic reconstruction tools Genome Biol 20 158 ),即AuReMe、CarveMe、Merlin、MetaDraft、ModelSEED、Pathway tools和RAVEN。Mendoza等在18个具体标准的基础上得出结论,每个工具在不同的方面显示出优势和不足,比较标准之一是软件的能力提供了一个现成的模型作为输出,使用是指可能执行的通量平衡分析(FBA)或FBA derived仿真技术预测生物代谢生理,包括生物质生产,在一个给定的化学环境。只有CarveMe和ModelSEED完全满足这个标准。
MetaGEM实战
本文主要介绍MetaGEM工具的使用。
官方链接: https://github.com/franciscozorrilla/metaGEM.
简介
一个易于使用的工作流程,用于生成特定环境的基因组尺度代谢模型,并直接从宏基因组数据预测微生物群落内的代谢相互作用。(Zorrilla F , Patil K R , Zelezniak A . metaGEM: reconstruction of genome scale metabolic models directly from metagenomes[J]. Nucleic Acids Research, 2021.)
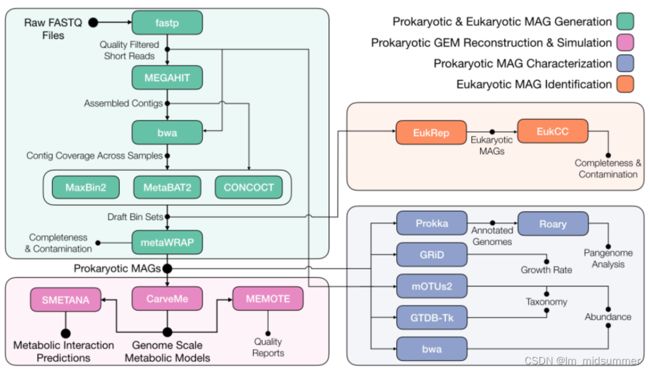

整个流程使用Snakemake实现,从原始的宏基因组的fastq文件开始,质控、组装、估计contig覆盖率、binning、Bin的改进和重组、MAG丰度定量和物种分类、CarveMe进行基因组规模代谢模型重建及质量报告,SEMATANA模拟重建的基因组规模代谢模型的肠道微生物群落。
安装
Clone the metaGEM repo and run automated setup script:
git clone https://github.com/franciscozorrilla/metaGEM.git
cd metaGEM
bash env_setup.sh
Load conda and activate environment:
module load anaconda3 # The exact module name will depend on the version installed on your cluster
source activate metagem
使用
以下教程全部来自于https://github.com/franciscozorrilla/unseenbio_metaGEM
总览:
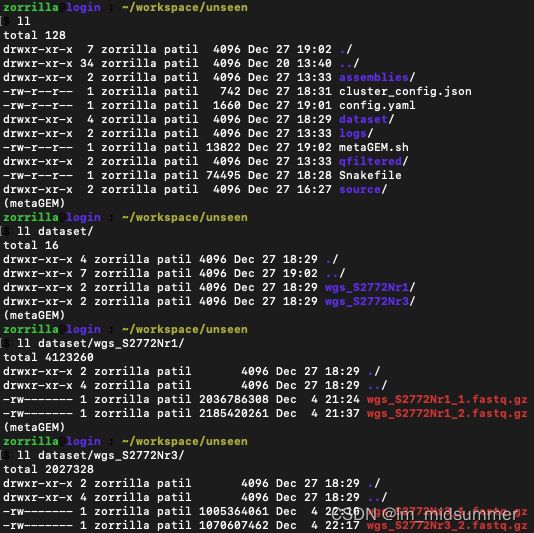
在dataset文件夹中的子目录中存放paierd-end的fastq数据,如下所示。MetGEM 将基于dataset文件夹中存在的子文件夹读取示例 ID,并将这些 ID 提供给 Snakefile 作为作业提交的通配符。
1. 使用fastp质量过滤reads
每个样本提交一个质量过滤工作,每个过滤工作有2个CPU和20GB 内存,最大运行时间为2小时
bash metaGEM.sh -t fastp -j 2 -c 2 -m 20 -h 2
可视化质量筛选结果:
bash metaGEM.sh -t qfilterVis
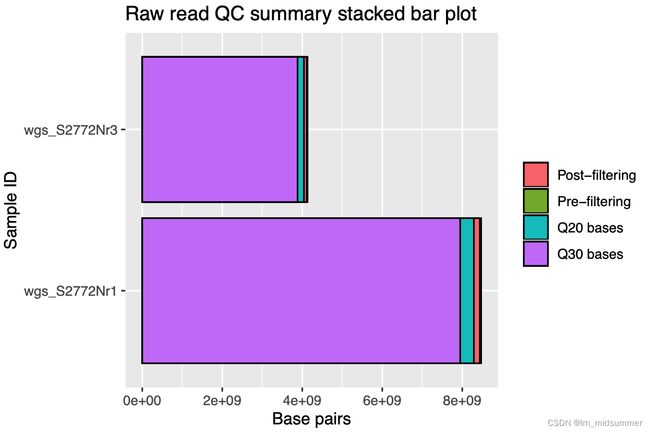
上述条形图显示样品 wgs_S2772Nr1具有大约40亿个碱基对,而样品 wgs_S2772Nr3具有 80亿个碱基对。两个样本都有良好的质量评分,大约98%的读数/碱基对在质量过滤后被保留。
2. 用 Megahit 组装
每个样品提交一个组装作业,每个组装作业有24个CPU和120GB 内存,最大运行时间为24小时:
bash metaGEM.sh -t megahit -j 2 -c 24 -m 120 -h 24
可视化组装结果:
bash metaGEM.sh -t assemblyVis
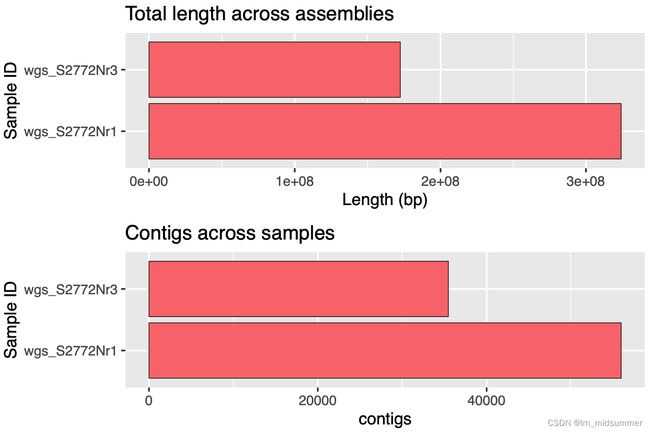
上述条形图显示,样品 wgs_S2772Nr1的组装包含约3.24亿个碱基对跨越约5.6万个contigs (平均约5.8 kbp/contig) ,而样品 wgs_S2772Nr3包含约1.72亿个碱基对跨越约3.5万个contigs(平均约4.9 kbp/contig)。这些contigs应该可以很好的组装。
3. 使用 CONCOCT、 MaxBin2和 MetaBAT2分箱
使用 bwa 和 samtools,将每组成对的末端读数与每组组装的组合进行交叉映射,以获得样品间组合的丰度/覆盖率。每个样本提交一个作业,每个作业有24个CPU和120GB 内存,最大运行时间为24小时:
bash metaGEM.sh -t crossMapSeries -j 2 -c 24 -m 120 -h 24
注意: 旧的 rule CrosMap 被分成了 CrosMapSeries 和 rosMapParaller 两部分。运行序列映射更加简单,但是从计算资源的角度来看,对于有大量样本的数据集,例如 N = 1000,运行映射的代价可能会高得令人望而却步。
在样本之间运行每个binners,使用连续覆盖率:
bash metaGEM.sh -t concoct -j 2 -c 24 -m 80 -h 10
bash metaGEM.sh -t metabat -j 2 -c 24 -m 80 -h 10
bash metaGEM.sh -t maxbin -j 2 -c 24 -m 80 -h 10
4. 使用 metWRAP 改进和重新组装
优化和重组bins
bash metaGEM.sh -t binRefine -j 2 -c 24 -m 150 -h 24
bash metaGEM.sh -t binReassemble -j 2 -c 24 -m 150 -h 24
可视化输出:
bash metaGEM.sh -t binningVis

我们可以看到样品 wgs_S2772Nr1和 wgs_S2772Nr1分别产生了88和47个bins,总共135个bins。虽然 CONCOCT 在产生的bins数量方面往往优于 MetaBAT2和 MaxBin2(特别是当有更多样本可用于利用 Contig 覆盖率信息时) ,但在这种情况下,MetaBAT2产生的bins最多。这突出了 metGEM 装箱实现的强度和灵活性,其中来自多个装箱的draft bins被改进和重新组装,以获得最佳的分箱(平均完整性86.5% 和平均污染1.2%) ,而不是依赖于单一的分箱方法。事实上,CONCOCT 倾向于生成完成度较高的bins(平均完成度89.2% ,平均污染1.9%) ,而 MetaBAT2倾向于生成污染较低的bins(平均完成度83.4% ,平均污染1.6%)。虽然 MaxBin2产生的bins比上图所示的多,但由于高污染,大多数bins不符合中等质量标准,即完整性 > 50% ,污染小于5% 。此外,与 CONCOCT (平均大小为2.45 Mbp & 287.5 contigs)或 MetaBAT2(平均大小为2.38 Mbp & 167.5 contigs)相比,metWRAP 重组bins改善了bins的连续性(平均大小为2.45 Mbp & 平均 contigs 161.9)。
5. 用 GTDB-tk 进行分类
首先让我们从 metWRAP 重组输出中提取我们的 DNA bins:
bash metGEM.sh -t extractDnaBins
运行 GTDB-tk 进行分类学分类:
bash metaGEM.sh -t gtdbtk -j 2 -c 24 -m 80 -h 12
在计算相对丰度之后,我们将可视化分类注释。
6. bwa 和 samtools 计算相对丰度
bash metaGEM.sh -t abundance -j 2 -c 24 -m 80 -h 12
可视化分类和相对丰富度:
bash metaGEM.sh -t compositionVis

从上图中排除的是具有未定义的物种水平分类的基因组的相对丰度,占样品 wgs_S2772Nr1和 wgs_S2772Nr3的相对丰度的24.9% 和12.7% 。
我们还可以在 R 中进行一些额外的手工分析,以检查样本之间个体物种丰度的相关性:
taxab %>% select(user_genome,sample,species,rel_ab) %>%
group_by(species) %>%
mutate(count=n()) %>%
filter(count==2) %>%
select(-count,-user_genome) %>%
pivot_wider(values_from = rel_ab,names_from = sample) -> scatter_dat
rval=cor(scatter_dat$wgs_S2772Nr1,scatter_dat$wgs_S2772Nr3)
lm_fit = summary(lm(wgs_S2772Nr1~wgs_S2772Nr3,data=scatter_dat))
pval=lm_fit$coefficients[8]
ggplot(scatter_dat,aes(x=wgs_S2772Nr1,y=wgs_S2772Nr3)) +
geom_point() +
geom_smooth(method = "lm") +
annotate("text", label = paste0("Pearson's r = ",signif(rval,digits = 4)), x = 0.015, y = 0.07,size=5) +
annotate("text", label = paste0("p-value = ",signif(pval,digits = 4)), x = 0.015, y = 0.06,size=5)

7. 利用 CarveMe 和 memote 重建和评估基因组尺度的代谢模型
尽管大多数 CarveMe 依赖项都是使用 metGEM conda recipie安装在 metGEM conda 环境中的,但是 CPLEX 解决方案要求用户在 IBM 注册以获得免费的学术许可证。
让我们从 metWRAP 重组输出中提取 ORF 注释的蛋白质bin,并将它们转储到一个单独的文件夹中,以便于并行工作提交:
bash metaGEM.sh -t extractProteinBins
默认情况下,模型将在全培养基上填充,但是这可以在 config.yaml 文件中轻松地进行调整。还可以向 media_db.tsv添加自定义培养基。Tsv 文件,它使用 BiGG 数据库代谢物 ID。现在让我们在生成的蛋白质bins上运行 CarveMe。请注意,metGEM 将从 protein_bins 文件夹中读取 MAG ID,也就是提取的 Protein Bins 规则存放蛋白质 bin 文件的位置。在这种情况下,我们将提交135个作业(每个 MAG 一个) ,每个作业有4个内核和20GB 内存,最大运行时间为4小时:
bash metaGEM.sh -t carveme -j 135 -c 4 -m 20 -h 4
生成模型后,可以使用 memte 对它们进行评估:
bash metaGEM.sh -t memote -j 135 -c 4 -m 20 -h 2
Memete 为每个模型生成详细的报告,还可以使用脚本 cli.py 对这些报告进行汇总和分析。我们可以用 metGEM 快速显示代谢物、基因和模型反应的数量:
bash metaGEM.sh -t modelVis
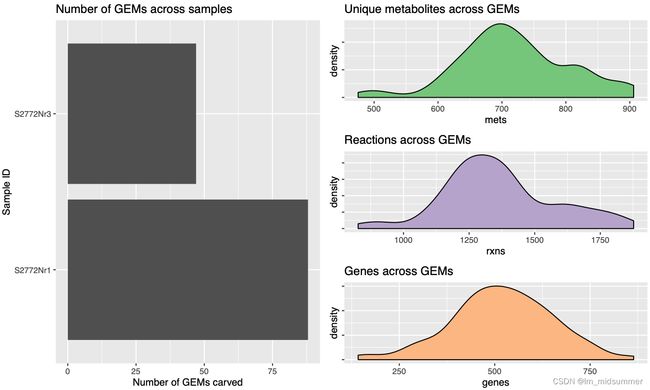
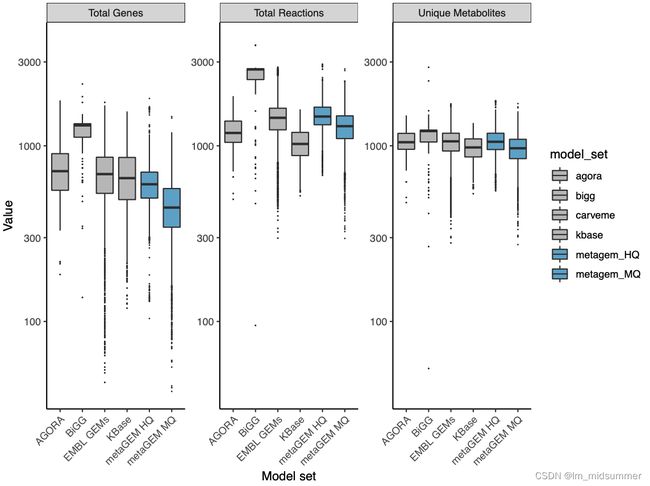
与我们出版的其他模型相比,这些模型具有相似的代谢物、反应和基因分布。
8. 使用SMETANA物种代谢偶联分析
首先,让我们将模型组织到示例特定的子目录中,以便于提交作业:
bash metaGEM.sh -t organizeGEMs
SMTANA 算法计算物种耦合评分(SCS) ,代谢物摄取评分(MUS)和代谢物产生评分(MPS) ,将其相乘以获得每个物种对之间每种可能的相互作用的 SMTANA 评分。欲了解更多关于实施和解释上述模拟的信息,请参阅原始文献。
我们可以通过修改 config.yaml 文件轻松地为计算实验配置模拟培养基。默认情况下,metaGEM 将模拟基础 media_db.tsv 中所有培养基中的社区。同样,可以通过展开 media_db.tsv 创建自定义模拟培养基,基于 BiGG 数据库代谢产物 ID。要注意,大型社区可能需要很长的运行时间才能完成,特别是如果提供了许多模拟培养基(社区模拟是按照每个培养基串行运行的)。
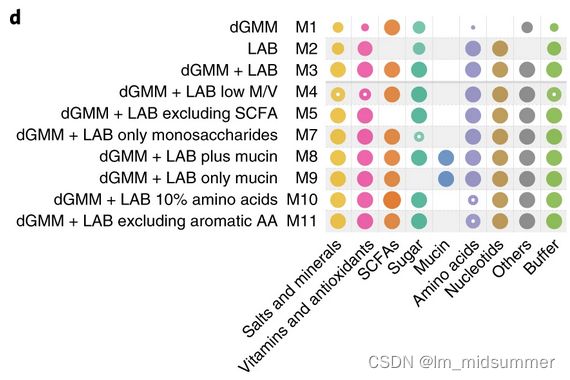
在这个演示中,将设置与在 metGEM 论文中相同的计算实验,其中模型填充在完全培养基(M3)上,并在没有芳香族氨基酸(M11)的介质中进行模拟。培养基组成由这篇文章提供,如下图:
bash metaGEM.sh -t smetana -j 2 -c 24 -m 40 -h 24
该输出文件包含模拟社区成员之间预测的新陈代谢相互作用:
community medium receiver donor compound scs mus mps smetana
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.33.p M_acald_e 0.0425531914893617 0.12 1 0.005106382978723404
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.33.p M_ala__D_e 0.0425531914893617 0.23 1 0.009787234042553192
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.33.p M_alaala_e 0.0425531914893617 0.02 1 0.000851063829787234
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.33.p M_phe__L_e 0.0425531914893617 1.0 1 0.0425531914893617
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.19.s M_acald_e 0.02127659574468085 0.12 1 0.002553191489361702
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.19.s M_ala__D_e 0.02127659574468085 0.23 1 0.004893617021276596
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.19.s M_alaala_e 0.02127659574468085 0.02 1 0.000425531914893617
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.40.s M_acald_e 0.02127659574468085 0.12 1 0.002553191489361702
all M11 wgs_S2772Nr3_bin.50.p wgs_S2772Nr3_bin.40.s M_ala__D_e 0.02127659574468085 0.23 1 0.004893617021276596
可视化来自样本 wgs_S2772Nr3的 SMETANA 模拟结果,可以从绘制社区内最常见的交换化合物的箱形图开始:
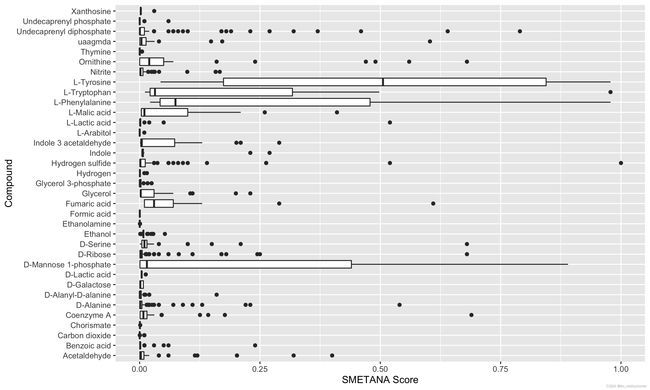
正如预期的那样,我们看到tryptophan, tyrosine, 和phenylalanine在这个肠道微生物群落中交换。我们也可以根据贡献者和接受者的分数来分组以识别关键种:
上图的顶部子图表明,Parasutterella excrementihominis 和 Agathobaculum butyriciproducens是关键的供体物种,尽管它们分别占群落相对丰度的0.61% 和0.97% 。另一方面,底部子图表明,Faecalicatena torques 和 Bacteroides salyersiae是最依赖于其他群落成员。有趣的是,我们还发现群落中数量最多的Faecalibacterium prausnitzii K物种完全依赖于Agathobaculum butyriciproducens产生的硫化氢(即这种相互作用的 SMETANA 得分为1)。
参考:
1 Yu Chen, Feiran Li, Jens Nielsen, Genome-scale modeling of yeast metabolism: retrospectives and perspectives, FEMS Yeast Research, Volume 22, Issue 1, 2022, foac003
2 Mendoza, S. N., Olivier, B. G., Molenaar, D. & Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol 20, 158 (2019).
3 刘立明,陈坚.基因组规模代谢网络模型构建及其应用[J].生物工程学报,2010,26(09):1176-1186.DOI:10.13345/j.cjb.2010.09.011.
4 Magnúsdóttir S, Heinken A, Kutt L, Ravcheev D A, Bauer E, Noronha A, Greenhalgh K, Jäger C, Baginska J, Wilmes P, Fleming R M T and Thiele I 2017 Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota Nat Biotechnol 35 81–9
5 Hamilton J J and Reed J L 2014 Software platforms to facilitate reconstructing genome-scale metabolic networks Environ Microbiol 16 49–59
6 Francisco Zorrilla, Filip Buric, Kiran R Patil, Aleksej Zelezniak. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes. Nucleic Acids Research, Volume 49, Issue 21, 2 December 2021, Page e126, https://doi.org/10.1093/nar/gkab815