ATAC-seq数据分析(一)
1 .下载需要的SRR数据
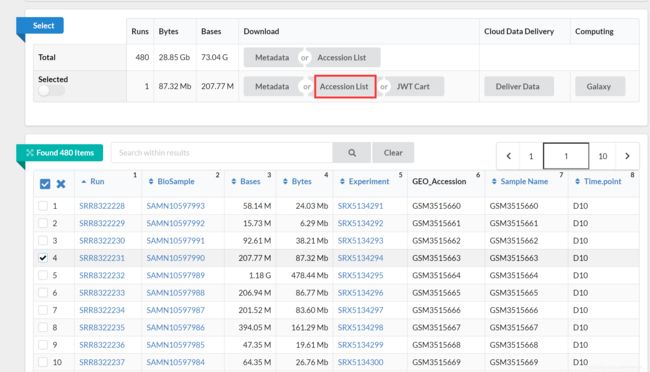
选择符合要求的数据(具体要看论文,这篇论文就提到了有的ATAC数据和RNA数据没有pass),选择完数据后点击Accession List,会得到SRR_Acc_List.txt
2 .使用sratoolkit(NCBI有安装教程)批量下载SRR数据
prefetch --option-file SRR_Acc_List.txt --output-directory /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/raw_data
#在SRR_Acc_List.txt所在的目录下执行。
这步执行完,若是创建了文件夹包含.sra文件,就参考我之前的博客,删除文件夹,留下.sra文件。
3 .判断文件是否文件少于样本数(此论文提到pass的SRR一共273个)
ls -l|grep "^-"| wc -l
激活处理ATAC数据的虚拟环境,下载需要的包
4 .批量将.sra文件转为fastq文件(bash)
touch fastq_demo.sh
chmod +x fastq_demo.sh
vim fastq_demo.sh
#!/bin/bash
cat SRR_Acc_List.txt | while read id;
do echo $id
cd raw_data/
if [-e $id.sra ];then
nohup fastq-dump -A $id -O /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/fastq_data --gzip --split-3
/home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/raw_data/$id.sra &
else
echo "No Files"
fi
cd ..
done
--split-3参数代表着如果是单端测序就生成一个 .fastq文件,如果是双端测序就生成_1.fastq 和*_2.fastq 文件 在论文和NCBI中都提到了这个数据集是双端测序 -A 命名文件 -O 存储位置
source fatsq_demo.sh
5 .使用trim_galor进行对fastq文件批量质量控制
cd fastq_data
ls *_1.fastq.gz > ls_1.txt
touch trim_demo.sh
chmod +x trim_demo.sh
vim trim_demo.sh
#!/bin/bash
cat ls_1.txt | while read i
do
echo $i
i=${i%_1.fastq.gz}
echo $i
nohup trim_galore --phred33 -q 25 --length 35 --stringency 4 -e 0.1 --fastqc --paired -o /home/yxchen/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/clean_fastq_data ${i}_1.fastq.gz ${i}_2.fastq.gz &
done
source trim_demo.sh
处理完可以再检查一下clean_fastq_data中的文件数量对不对
6 .质控的可视化(对比trim前后的fastq.gz的FastQC report,主要看adapter Content)
fastqc -t 5 -O /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/qc_fastq /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/fastq_data/*.fastq.gz
fastqc -t 5 -O /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/qc_val_fastq /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/clean_fastq_data/*.fq.gz
7 .bowtie2序列比对
conda install bowtie2我失败了,我不经过conda下载bowtie2:
wget https://sourceforge.net/projects/bowtiebio/files/bowtie2/2.3.5.1/bowtie2-2.3.5.1-linux-x86_64.zip/download
unzip bowtie2-2.3.5.1-linux-x86_64.zip
vim ~/.bashrc
#bowtie2
export PATH="/home/***/bowtie2/bowtie2-2.3.5.1-linux-x86_64/:$PATH"#添加路径
source ~/.bashrc
下载人类参考基因组hg19(http://www.bio-info-trainee.com/1985.html):
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz #九百多兆
tar zvfx chromFa.tar.gz
cat *.fa > hg19.fa
rm chr*.fa
rm chromFa.tar.gz
对参考文件建立索引(时间比较长)
bowtie2-build /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/reference/hg19/hg19.fa hg19_genome
或者直接下载人类基因组索引文件(推荐)
wget ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/hg19.zip
批处理比对全流程,去除PCR重复和高质量的比对结果,最后转为.bed文件
#!/bin/bash
bowtie2_index='/home/yxchen/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/hg19_genome/hg19'
#bowtie2_index='/home/yxchen/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/reference/hg19/hg19_index/hg19_genome'
cat ls_1.txt | while read id
do
#echo $id
id=${id%_1_val_1.fq.gz}
#echo $id
fq1=${id}_1_val_1.fq.gz
fq2=${id}_2_val_2.fq.gz
id=${id%.sra}
#echo $id
#echo $fq1
#echo $fq2
bowtie2 -p 5 --very-sensitive -X 2000 -x $bowtie2_index -1 $fq1 -2 $fq2 | samtools sort -O bam -@ 5 -o -> /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.bam
samtools index /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.bam
samtools flagstat /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.bam > /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.raw.stat
gatk MarkDuplicates -MAX_FILE_HANDLES 1000 -I /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.bam -O /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.rmdup.bam --REMOVE_DUPLICATES true -M test.log
samtools index /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.rmdup.bam
samtools flagstat /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.rmdup.bam > /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.rmdup.stat
samtools view -h -f 2 -q 30 /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.rmdup.bam |grep -v chrM | samtools sort -O bam -@ 5 -o ->/home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.last.bam
samtools index /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.last.bam
samtools flagstat /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.last.bam > /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.last.stat
bedtools bamtobed -i /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.last.bam > /home/***/JupyterNotebook/Multiomics_data/Parallel/boold_ATAC/alignment/${id}.bed
done
bowtie2_index基因索引文件的位置要注意,不仅仅是要写上存放的位置,还要标注上基因索引文件的类型名字:如下:
pwd显示的是我的参考基因组的索引存储的位置,但是bowtie2_index不能只写pwd的位置信息,还要加上ls出来的索引文件的类型名,这里就是加上hg19。否则会报错,does not exist or is not a Bowtie 2 index Exiting now …![]()
各个数据格式的补充信息
** ‘.sra’的后缀是SRA数据库对fastq文件的特殊压缩。使用前,我们需要将其解压为fastq文件。简单来说,测序得到的是带有质量值的碱基序列(fastq格式),参照基因组是(fasta格式),用比对工具把fastq格式的序列回帖到对应的fasta格式的参考基因组序列,就可以产生sam格式的比对文件。 把sam格式的文本文件压缩成二进制的bam文件可以节省空间,如果对参考基因组上面的各个区段标记它们的性质,比如哪些区域是外显子,内含子,UTR等等,这就是gtf/gff格式。 如果只是为了单纯描述某个基因组区域,就是bed格式文件,记录染色体号以及起始终止坐标,正负链即可。 如果是记录某些位点或者区域碱基的变化,就是VCF文件格式(Variant Call Format(VCF)是一个用于存储基因序列突变信息的文本格式。 可以表示单碱基突变, 插入/缺失, 拷贝数变异和结构变异等。 通常是对BAM文件格式的比对结果进行处理得到的。 BCF格式文件是VCF格式的二进制文件)。**
生物技能树_数据存放类型