深度学习预测酶活性参数提升酶约束模型构建从头环境搭建
前言
这项工作开发了一种用深度学习来预测酶活性参数的方法(DLKcat),主要采用了针对底物的图神经网络和针对蛋白质的卷积神经网络。通过从公开的数据库中获取和数据预处理,最终获得了超过一万六千条高质量的数据。随后,该团队在此数据基础上训练了深度学习的模型,这个模型不仅能够很好的预测整体数据集和测试数据集。更值得一提的是,即使对于测试数据集中那些蛋白或者底物没有出现在训练数据集中的数据,模型也有很好的预测能力。同时,这套深度学习模型也能够精准的预测酶的多功能性。除此以外,模型也能够捕捉酶的某些氨基酸突变所带来的酶活性的影响,对于野生型的酶及其突变体都有相当不错的预测能力。为了进一步解释深度学习网络模型,该工作采用了注意力机制从神经网络中得到重要的信号并对其可视化,进一步揭示了哪些氨基酸残基对酶的活性有重要的影响。
1.安装分为两部分
DLKcat工具箱是一个Matlab/Python软件包,用于预测kcats和生成ecGEMs。该程序包分为两部分。DeeplearningApproach和BayesianApproach。DeeplearningApproach提供了一个基于深度学习的kcat预测工具,而BayesianApproach提供了一个基于贝叶斯的自动管道,使用预测的kcats构建ecModels。
下面均以win10系统操作为主,linux和MACos有细微区别

第一部分:DeeplearningApproach环境配置
1.下载DLKcat package
git clone https://github.com/SysBioChalmers/DLKcat
2.下载依赖包
pip install numpy requests torch torchvision rdkit-pypi sklearn
3.进入DeeplearningApproach目录
cd DLKcat/DeeplearningApproach
4.在Data文件夹下解压input.zip
unzip Data/input.zip
5.在DLKcat文件夹下进入Code/example。
cd Code/example
6.运行
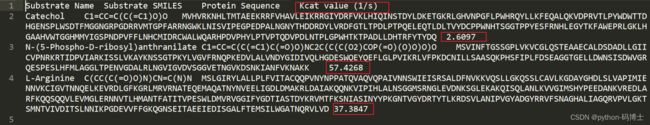
现在可以通过一个命令行使用训练好的深度学习模型进行预测。在这里,需要准备一个输入文件,可以查看Code/example/input.tsv。对于输入文件,应该提供蛋白质序列,用户还需要提供底物(化合物)名称或底物(化合物)SMILES,但推荐底物SMILES。如果很难找到底物SMILES,请提供底物名称,底物SMILES留空。
如图:大致就是这样的文件,这里是预测了3种底物与化合物的Kcat值

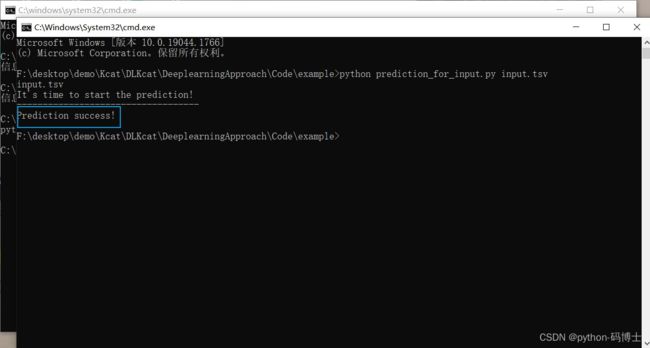
然后预测结果(output.tsv文件)将在Code/example目录下输出。
如图:预测出了Kcat值
7.如果想要分析并重新生成数据
要重新生成所有数据,解压Data/input.zip中的input.zip文件,并在Code/analysis目录中运行相应的数据函数。
8.预处理过程
BRENDA数据库的数据收集和清理。
brenda_retrieve.py :访问网络客户端并从BRENDA数据库中检索数据集。
brenda_download.py:读取检索到的文件中的所有数据并输出所有EC文件
findMaxKvalues_AllOrgs.py 读取所有的EC文件并找出所选微生物的每个底物的最大值。
brenda_kcat_preprocess.py,将所有EC文件中的Kcat数据生成为一个文件。
brenda_kcat_clean.py 从BRENDA数据库中清理数据集。
brenda_sequence.py 从BRENDA数据库中获取蛋白质序列的一个例子
brenda_sequence_organism.py 获得所有基于EC号和生物体的数据的蛋白质序列,并输出到文件夹中供下一步使用。
从SABIO-RK数据库收集和清理数据
sabio_download.py以获得对网络客户端的访问并从SABIO-RK数据库中下载数据集。
sabio_kcat_unisubstrate.py,从下载的文件中读取所有数据,并输出到一个文件中供进一步使用
sabio_kcat_clean_unisubstrate.py,通过统一所有条目来清理数据。
sabio_kcat_clean.py,用于清理SABIO-RK的数据。
sabio_kcat_unisubstrate_mutant.py来注释酶的类型信息,即野生型或突变型。
uniprot_sequence.py,通过uniprot蛋白质ID获得蛋白质序列。
sabio_get_smiles.py,根据SABIO-RK数据的底物名称获得规范的SMILES,并输出一个文件供使用。
从BRENDA和SABIO-RK数据库获得的数据集的数据组合。
combination_brenda_sabio.py 初步结合BRENDA和SABIO-RK数据库中的Kcat数据。
combination_database_data.py,将所有合并的数据生成一个文件,用于深度学习和进一步分析。
9.注释
通过Code/model目录查看深度学习的构建和评估
通过深度学习模型对343种酵母菌/真菌进行预测。
要获得基于训练好的深度学习模型的343种酵母菌/真菌的预测结果,解压Data/input.zip中的input.zip文件,并运行Code/rediction目录中的相应函数即可
第一部分我已实现
第二部分:贝叶斯搭建(这一部分遇到一些问题,如果有搞好的,请艾特我一下)
1. 首先介绍一下搭建需要的工具
MATLAB 9.1 (R2016b) 或者更高级版本 + Optimization Toolbox.
MATLAB
matrix&laboratory两个词的组合,意为矩阵工厂(矩阵实验室),软件主要面对科学计算、可视化以及交互式程序设计的高科技计算环境。它将数值分析、矩阵计算、科学数据可视化以及非线性动态系统的建模和仿真等诸多强大功能集成在一个易于使用的视窗环境中,为科学研究、工程设计以及必须进行有效数值计算的众多科学领域提供了一种全面的解决方案,并在很大程度上摆脱了传统非交互式程序设计语言(如C、Fortran)的编辑模式。
Optimization Toolbox
提供各种函数,可用于求最小化或最大化目标且满足约束的参数。该工具箱包含用于线性规划 (LP)、混合整数线性规划 (MILP)、二次规划 (QP)、二阶锥规划 (SOCP)、非线性规划 (NLP)、约束线性最小二乘、非线性最小二乘和非线性方程的求解器。
COBRA toolbox for MATLAB.
基于约束的重建和分析工具箱
值得注意的是,安装cobratoolbox时要满足以下环境
查看指南,正确配置系统。
确保安装一个兼容的解算器。在这里检查兼容性。
可以按照这些详细说明来安装TOMLAB、IBM ILOG CPLEX、GUROBI或MOSEK。
这些都叫做替代求解器、
这也叫免费?还费用不高
安装主要分为五部(最好有教育邮箱,可以白嫖一段时间的软件使用权)
1.下载软件
2.购买证书(教育邮箱白嫖)
3.将证书放到指定位置
4.设置环境变量
5.测试安装是否成功
在MATLAB上运行>> testAll
接下来就是安装环节
1.下载资源(在终端运行)
git clone --depth=1 https://github.com/opencobra/cobratoolbox.git cobratoolbox
2.在MATLAB上进入cobratoolbox文件夹并运行>> initCobraToolbox
RAVEN toolbox for MATLAB.
RAVEN(代谢网络的重建、分析和可视化)工具箱2是Matlab的一个软件套件,允许半自动重建基因组尺度的模型(GEMs)。它利用已发表的模型和/或KEGG、MetaCyc数据库,加上广泛的填空和质量控制功能。该软件套件还包含了模拟结果和全向数据的可视化方法,以及一系列执行模拟和分析结果的方法。该软件是一个有用的工具,用于代谢背景下的全系统数据分析和基于蛋白质同源性的代谢网络的精简重建。
libSBML MATLAB API (推荐5.17.0版本)
LibSBML是一个免费的、开源的编程库,帮助读取、写入、操作、翻译和验证SBML文件和数据流。它本身并不是一个应用程序(尽管它确实带有示例程序),而是一个可以嵌入到自己的应用程序中的库。
接下来是安装环境
第一种安装方法:
1.在本地计算机解压文件
2.打开MATLAB,将该文件添加到MATLAB路径
3.验证是否正常
在MATLAB中切换到该文件夹,输入TranslateSBML('test.xml'),最终输出一个测试模型的matlab结构
4.用savepath命令将这些操作保存到MATLAB中
第二种安装方法:
1.在本地计算机解压文件
2.打开MATLAB,进入到该文件夹目录并运行installSBML
3.验证如上
Violin Plots for Matlab
小提琴图是箱形图的一个易于阅读的替代品,它用数据的核密度估计值代替了箱形图,并可选择叠加数据点本身。原有的boxplot形状仍然作为灰色框/线包括在小提琴的中心。
pval_adjust
MATLAB/Octave用于调整多重比较的p值的函数。给定一组p值,返回用几种方法之一调整的p值。这是p.adjust R函数的一个实现,该函数的文档可以在http://www.inside-r.org/r-doc/stats/p.adjust查看。
在贝叶斯过程中,使用了IBM CPLEX 12.10,在调用函数solveModel时将ibm_cplex替换成gurobi,调整函数abc_matlab_max.m后也可以使用gurobi。
对于其他过程,将使用cobra工具箱中的默认解算器,IBM CPLEX 12.10或gurobi都可以。
2.使用
1.将DLKcat加入到MATLAB环境变量中。
2.如果只想运行部分过程,可以从zenode链接中下载原始结果,并将其与结果文件夹结合起来。
3.使用深度学习模型提取信息用于kcat预测
该函数是在集群上运行的,调整unction initcluster.m来设置集群路径。
调整函数ForKcatPrediction/WriteFile_pre_cluster.m,用于调用函数ForKcatPrediction/writeFileForKcatPrediction.m,提取深度学习模型预测所需的信息,并为每个模型生成一个txt文件。模型文件来自Yeast-Species-GEMs
4.对于重建酶制约的模型。酶约束模型可以像任何代谢模型一样,使用COBRA或RAVEN等工具箱。
1.运行深度学习步骤来预测Kcat。所有预测的kcat文件都可以在zenode链接文件夹中找到,请从zenode链接中下载原始结果,并与结果文件夹结合。
2. 在集群上运行的,要适应用initcluster.m这个命令来设置集群的路径。创建自己的bash文件来在这个集群上运行作业。
3.运行classicDLModelGeneration_cluster.m来收集包含kcat和蛋白质信息的酶数据,并生成Classical-ecGEM和DL-ecGEM。物种应按Strain.txt中的索引顺序输入。
4.运行BayesianModelGeneration_cluster.m获得后验均值ecGEM。物种应按Strain.txt中的索引顺序输入。
5.运行getEmodel.m,从enzymedata和GEM中获得GECKO版本的ecGEMs。之后,所有的GECKO函数都可以用于进一步分析。所有的ecGEMs都存储在zenode链接中。
5.运行酶约束模型的分析并重现所有特征
要重现所有的特征,运行相应的特征函数,请从zenode链接中下载原始结果并与结果文件夹结合。
注释
如果需要343种酵母菌/真菌的ecGEMs,在文件夹中找到ecGEMs:在zenode link中的Results/ecGEMs. 每个物种有三个版本的ecGEMs可供选择。推荐使用Posterior_mean-ecGEMs版本。
最后介绍一下集群这个概念
集群就是指一组(若干个)相互独立的计算机,利用高速通信网络组成的一个较大的计算机服务系统,每个集群节点(即集群中的每台计算机)都是运行各自服务的独立服务器。这些服务器之间可以彼此通信,协同向用户提供应用程序,系统资源和数据,并以单一系统的模式加以管理。当用户请求集群系统时,集群给用户的感觉就是一个单一独立的服务器,而实际上用户请求的是一组集群服务器。