RDkit入门
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档
文章目录
- 前言
- 一、读写分子
-
- 1.1 读取分子
- 1.2 写入分子
- 二、操作分子
-
- 2.1 获取分子中的原子 GetAtoms()
- 2.2 获取分子中的键 GetBonds()
- 2.3 根据原子编号获取键 GetAtomWithIdx()
- 2.4 获取原子的坐标信息
- 2.5 获取原子信息
- 2.6 根据键的编号获取键 GetBondWithIdx( )
- 2.7 获取键的信息
- 2.8 获取分子中所有的环 GetSymmSSSR(m)
- 2.9 获取分子中环的个数 GetSSSR(m)
- 2.10 获取分子中环的信息 GetRingInfo()
- 三、修改分子
-
- 3.1 增删H原子
- 3.2 芳香共轭键和库里单双键Chem.Kekulize(m)
- 四、处理2D分子
-
- 4.1 产生2D坐标AllChem.Compute2DCoords(m)
- 4.2 基于3D构象产生2D取向 AllChem.GenerateDepictionMatching3DStructure()
- 五、处理3D分子
-
- 5.1 产生3D构象 AllChem.EmbedMolecule(m)
- 5.2 产生多个3D构象
- 5.3 计算构象的RMSD
- 5.4 MMFF力场对构象进行优化AllChem.MMFFOptimizeMoleculeConfs
- 六、保存分子对象
- 七、绘制分子【可视化分子】
-
- 7.1 分子对象转化为图片
- 7.2 显示分子中原子的编号
- 7.3 多个分子按照grid显示
- 7.4 多个分子基于公共骨架按照grid显示
- 八、子结构搜索
-
- 8.1 判断是否有子结构m.HasSubstructMatch(patt)
- 8.2 获取第一个子结构对应的原子编号m.GetSubstructMatch()
- 8.3 获取对应所有子结构(m.GetSubstructMatches)对相应的原子编号
- 8.4 子结构搜索考虑手性
- 九、化学转换
-
- 9.1 删除子结构 AllChem.DeleteSubstructs(m,patt)
- 9.2 取代基替换 AllChem.ReplaceSubstructs(m,patt,repl)
- 9.3 SAR分析——core可视化Chem.ReplaceSidechains(m1,core)
- 9.4 SAR分析——sidechain可视化Chem.ReplaceCore(m1,core)
- 9.5 拆分片段 Chem.GetMolFrags(tmp,asMols=True)
- 9.6 对多个分子进行SAR分析(待完成)
- 9.7 获取分子骨架Murcko Decomposition
- 9.8 最大公共子结构FindMCS
- 十、指纹和相似性
-
- 10.1 拓扑指纹 Chem.RDKFingerprint(x)
- 10.2 MACCS 指纹MACCSkeys.GenMACCSKeys(mol)
- 10.3 Atom Pairs
- 10.4 topological torsions
- 10.5 摩根指纹(圆圈指纹)AllChem.GetMorganFingerprint(mol,2)
- 10.6 基于指纹计算相似性
-
- 10.6.1 10.6.1 基于拓扑指纹 和 Tanimoto相似性方法(默认) 指纹计算3个分子的相似性
- 10.6.2 基于MACCS 指纹和Dice 相似性方法计算相似性
- 10.7 摩根指纹的形式
- 10.8 摩根指纹->ECFP4 和 摩根指纹->FCFP4的比较
- 10.9 用户也可以自己定义atom invariants
- 10.10 解释摩根指纹中bit的含义
- 10.11 获取指纹对应的结构
- 10.12 可视化指纹中的bit
- 10.13 可视化摩根指纹中的bit
- 10.14 可视化拓扑指纹中的bit
- 10.15 基于分子指纹挑选差异较大的分子
- 10.16 相似性地图
- 十一、分子性质(描述符)
-
- 11.1 性质描述符计算
- 11.2 计算分子的价电子数目
- 11.3 原子对性质的贡献可视化
- 总结
前言
今天开始写RDkit的入门教程,每天学习和写一小点,相信一句话“不积跬步无以至千里,不积小流无以成江海”,所以就慢慢来,一点一滴积累叭,从最简单的开始喽
一、读写分子
1.1 读取分子
#read smiles
smi='CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3'
from rdkit import Chem
from rdkit.Chem import AllChem
mol = Chem.MolFromSmiles(smi)
print(type(mol))
输出
#read mol
from rdkit import Chem
mol = Chem.MolFromMolFile('./ligand.mol')
print(type(mol))
输出
#read sdf
from rdkit import Chem
mols_suppl = Chem.SDMolSupplier('./ligand.sdf') # 划重点,和其他形式的逻辑不一样
print(type(mols_suppl))
输出
#more sdfs
mol_1= mols_suppl[0]
print(type(mol_1))
for mol in mols_suppl:
print(type(mol))
输出
#read mol2
from rdkit import Chem
mol2 = Chem.MolFromMol2File('./ligand.mol2')
print(type(mol2))
输出
#read protein DNA RNA
seq='GGGGG'
mol = Chem.MolFromSequence(seq)
smi = Chem.MolToSmiles(mol)
print("smi",smi)
输出smi NCC(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)O
#read pdb
pdb = Chem.MolFromPDBFile('./protein.pdb')
print(type(pdb))
输出
综上,rdkit中的Chem可以将别的形式的mol转化为rdkit形式的,再以此为中介转换为其它形式的
eg: mol2转pdb
mol2 = Chem.MolFromMol2File('./ligand.mol2')
pdb = Chem.MolToPDBBlock(mol2)
print(pdb,file=open('./ligand.pdb','w+'))
备注:目前RDkit还不支持转成mol2
1.2 写入分子
smi='CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3'
mol = Chem.MolFromSmiles(smi)
smi = Chem.MolToSmiles(mol)
print(smi)
molblock = Chem.MolToMolBlock(mol) # 转化为可保存的形式
print(molblock)
print(molblock,file=open('ligand.mol','w+')) # 写入文件,进行保存
输出为 CC©OC(=O)C©NP(=O)(OCC1OC(n2ccc(=O)[nH]c2=O)C©(F)C1O)Oc1ccccc1
RDKit 2D
36 38 0 0 0 0 0 0 0 0999 V2000
7.7598 -0.2393 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
8.6415 -1.4528 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
10.1333 -1.2960 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
8.0314 -2.8231 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
8.9131 -4.0367 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
10.4049 -3.8799 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
8.3030 -5.4070 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
9.1846 -6.6205 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
6.8112 -5.5638 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
6.2011 -6.9341 0.0000 P 0 0 0 0 0 0 0 0 0 0 0 0
5.5910 -8.3044 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
4.8308 -6.3240 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
4.6740 -4.8322 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
3.3037 -4.2221 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.0046 -4.9721 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.8899 -3.9684 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.5000 -2.5981 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.9918 -2.7549 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.7500 -1.2990 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.5000 2.5981 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.5000 0.0000 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.5000 -2.5981 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.0082 -5.1819 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.4091 -3.2184 0.0000 F 0 0 0 0 0 0 0 0 0 0 0 0
1.8478 -6.4639 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
7.5714 -7.5442 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
7.7282 -9.0360 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
6.5147 -9.9176 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
6.6715 -11.4094 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
8.0418 -12.0195 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
9.2553 -11.1379 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
9.0985 -9.6461 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
2 3 1 0
2 4 1 0
4 5 1 0
5 6 2 0
5 7 1 0
7 8 1 0
7 9 1 0
9 10 1 0
10 11 2 0
10 12 1 0
12 13 1 0
13 14 1 0
14 15 1 0
15 16 1 0
16 17 1 0
17 18 1 0
17 19 1 0
19 20 1 0
20 21 2 0
21 22 1 0
22 23 2 0
22 24 1 0
24 25 1 0
25 26 2 0
16 27 1 0
16 28 1 0
15 29 1 0
10 30 1 0
30 31 1 0
31 32 2 0
32 33 1 0
33 34 2 0
34 35 1 0
35 36 2 0
18 14 1 0
25 19 1 0
36 31 1 0
M END
二、操作分子
2.1 获取分子中的原子 GetAtoms()
from rdkit import Chem
smi='CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3'
mol = Chem.MolFromSmiles(smi)
atoms = mol.GetAtoms()
print(atoms)
print(type(atoms))
print(atoms[0])
print(type(atoms[0]))
输出为:
2.2 获取分子中的键 GetBonds()
from rdkit import Chem
smi='CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3'
mol = Chem.MolFromSmiles(smi)
bonds = mol.GetBonds()
print(bonds)
print(type(bonds))
print(bonds[0])
print(type(bonds[0]))
输出为:
2.3 根据原子编号获取键 GetAtomWithIdx()
from rdkit import Chem
smi='CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3'
mol = Chem.MolFromSmiles(smi)
atom0 = mol.GetAtomWithIdx(0)
print(atom0)
print(type(atom0))
输出为:
2.4 获取原子的坐标信息
from rdkit import Chem
mol= Chem.MolFromMolFile('./1a30_ligand.sdf',sanitize=False)
print(mol.GetConformer().GetAtomPosition(1)[0])
print(mol.GetConformer().GetAtomPosition(1).x)
print(mol.GetConformer().GetAtomPosition(1).y)
print(mol.GetConformer().GetAtomPosition(1).z)
x,y,z = mol.GetConformer().GetAtomPosition(1)
print('x:'+str(x)+' y:'+str(y)+' z:'+str(z))
xyz = list(mol.GetConformer().GetAtomPosition(1))
print(xyz)
print(mol.GetConformer().GetAtomPosition(0)[0])
print(mol.GetConformer().GetAtomPosition(0).x)
print(mol.GetConformer().GetAtomPosition(0).y)
print(mol.GetConformer().GetAtomPosition(0).z)
x,y,z = mol.GetConformer().GetAtomPosition(0)
print('x:'+str(x)+' y:'+str(y)+' z:'+str(z))
xyz = list(mol.GetConformer().GetAtomPosition(0))
print(xyz)
输出为:5.733
5.733
26.394
5.165
x:5.733 y:26.394 z:5.165
[5.733, 26.394, 5.165]
4.841
4.841
27.576
5.31
x:4.841 y:27.576 z:5.31
[4.841, 27.576, 5.31]
2.5 获取原子信息
‘GetAtomMapNum’: map id 原子smarts形式冒号后面的数字,如[N:4], map id 就是4。 ‘GetAtomicNum’: 获取原子对应的元素编号,如碳原子的AtomicNum就是6 ‘GetBonds’: 该原子参与形成的键的列表。 ‘GetFormalCharge’: 该原子的电荷 ‘GetChiralTag’:原子的手性信息 ‘GetDegree’: 原子的度,这里的度指的是这个原子参与形成键的数目 ‘GetIsAromatic’: 判断原子是否是芳香性原子 ‘GetIdx’: 获取原子的编号 ‘GetNeighbors’: 获取相连的原子列表 ‘GetSmarts’: 获取原子的Smarts形式 ‘GetSymbol’:获取原子的元素符号 IsInRing(): 判断原子是否在环上 IsInRingSize(n): 判断原子是否在n-元环上
2.6 根据键的编号获取键 GetBondWithIdx( )
from rdkit import Chem
smi='CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3'
mol = Chem.MolFromSmiles(smi)
bond1 = mol.GetBondWithIdx(1)
print(bond1)
print(type(bond1))
输出为:
2.7 获取键的信息
GetBondType():获取键的类型 ‘GetBeginAtom()’: 组成键的第一个原子 GetBeginAtomIdx(): 键的第一个组成原子编号 GetEndAtomIdx():键的第二个组成原子编号 ‘GetEndAtom()’: 组成键的第二个原子 ‘IsInRing()’: 判断是否在环上 ‘IsInRingSize(n)’:判断是否在n-元环上
from rdkit import Chem
m = Chem.MolFromSmiles('OC1C2C1CC2')
atom2 = m.GetAtomWithIdx(2)
print("atom2 in ring:",atom2.IsInRing())
print("atom2 in 3-ring:",atom2.IsInRingSize(3))
print("atom2 in 4-ring:",atom2.IsInRingSize(4))
print("atom2 in 5-ring:",atom2.IsInRingSize(5))
输出为:atom2 in ring: True
atom2 in 3-ring: True
atom2 in 4-ring: True
atom2 in 5-ring: False
2.8 获取分子中所有的环 GetSymmSSSR(m)
from rdkit import Chem
m = Chem.MolFromSmiles('OC1C2C1CC2')
ssr = Chem.GetSymmSSSR(m)
num_ring = len(ssr)
print("num of ring",num_ring)
for ring in ssr:
print("ring consisted of atoms id:",list(ring))
输出为:num of ring 2
ring consisted of atoms id: [1, 2, 3]
ring consisted of atoms id: [4, 5, 2, 3]
2.9 获取分子中环的个数 GetSSSR(m)
from rdkit import Chem
m = Chem.MolFromSmiles('OC1C2C1CC2')
num_ring = Chem.GetSSSR(m)
print("num rings:",num_ring)
输出为:num rings: 2
2.10 获取分子中环的信息 GetRingInfo()
from rdkit import Chem
m = Chem.MolFromSmiles('OC1C2C1CC2')
ri = m.GetRingInfo()
print(type(ri))
输出为:
三、修改分子
3.1 增删H原子
from rdkit import Chem
m = Chem.MolFromSmiles('OC1C2C1CC2')
m2 = Chem.AddHs(m)
print("m Smiles:",Chem.MolToSmiles(m))
print("m2 Smiles:",Chem.MolToSmiles(m2))
print("num ATOMs in m:",m2.GetNumAtoms())
print("num ATOMs in m2:",m.GetNumAtoms())
输出为:m Smiles: OC1C2CCC12
m2 Smiles: [H]OC1([H])C2([H])C([H])([H])C([H])([H])C12[H]
num ATOMs in m: 14
num ATOMs in m2: 6
3.2 芳香共轭键和库里单双键Chem.Kekulize(m)
m = Chem.MolFromSmiles('c1ccccc1')
for bond in m.GetBonds():
print(bond.GetBondType())
输出为:AROMATIC
AROMATIC
AROMATIC
AROMATIC
AROMATIC
AROMATIC
m = Chem.MolFromSmiles('c1ccccc1')
Chem.Kekulize(m)
for bond in m.GetBonds():
print(bond.GetBondType())
print("bond 1 is aromatic",m.GetBondWithIdx(1).GetIsAromatic())
print("atom 1 is aromatic",m.GetAtomWithIdx(1).GetIsAromatic())
输出为DOUBLE
SINGLE
DOUBLE
SINGLE
DOUBLE
SINGLE
bond 1 is aromatic True
atom 1 is aromatic True
四、处理2D分子
4.1 产生2D坐标AllChem.Compute2DCoords(m)
from rdkit.Chem import Draw
from rdkit import Chem
smis=[
'COC1=C(C=CC(=C1)NS(=O)(=O)C)C2=CN=CN3C2=CC=C3',
# 'CCN(CC1=C(C=CC(=C1)C(F)(F)F)C2=CC(=C3N2C=NC=C3)CC(=O)O)C(=O)C4CC4',
'C1=CC2=C(C(=C1)C3=CN=CN4C3=CC=C4)ON=C2C5=CC=C(C=C5)F',
'COC(=O)C1=CC2=CC=CN2C=N1',
'C1=C2C=C(N=CN2C(=C1)Cl)C(=O)O',
]
template = Chem.MolFromSmiles('c1nccc2n1ccc2')
AllChem.Compute2DCoords(template)
mols=[]
for smi in smis:
mol = Chem.MolFromSmiles(smi)
AllChem.GenerateDepictionMatching2DStructure(mol,template)
mols.append(mol)
img=Draw.MolsToGridImage(mols,molsPerRow=4,subImgSize=(200,200),legends=['' for x in mols])
img
输出为:
4.2 基于3D构象产生2D取向 AllChem.GenerateDepictionMatching3DStructure()
五、处理3D分子
5.1 产生3D构象 AllChem.EmbedMolecule(m)
mol=Chem.MolFromSmiles('c1nccc2n1ccc2')
m2=Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
m3 = Chem.RemoveHs(m2)
print(mol)
输出为:
5.2 产生多个3D构象
AllChem.EmbedMultipleConfs(m2, numConfs=10)
m2.GetConformer(1)
m2.GetConformers()
输出为:
5.3 计算构象的RMSD
rmslist = []
AllChem.AlignMolConformers(m2, RMSlist=rmslist)
rms = AllChem.GetConformerRMS(m2, 1, 9, prealigned=True)
print(rms)
print(rmslist)
0.02120375053020155
[0.028548924016513377, 0.04297710273927512, 0.0464421857004034, 0.030551642271433192, 0.029351858166554768, 0.041756778675049934, 0.02797758243996998, 0.056925146349127216, 0.0322206521175928]
5.4 MMFF力场对构象进行优化AllChem.MMFFOptimizeMoleculeConfs
res = AllChem.MMFFOptimizeMoleculeConfs(m2,numThreads=0)
六、保存分子对象
七、绘制分子【可视化分子】
7.1 分子对象转化为图片
opts = DrawingOptions()
opts.bondLineWidth=3
Draw.MolToFile(mol,'filename.png',options=opts)
7.2 显示分子中原子的编号
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole #Needed to show molecules
from rdkit.Chem.Draw.MolDrawing import MolDrawing, DrawingOptions #Only needed if modifying defaults
opts = DrawingOptions()
opts.includeAtomNumbers=True
m = Chem.MolFromSmiles('OC1C2C1CC2')
opts.includeAtomNumbers=True
opts.bondLineWidth=2.8
Draw.MolToImage(m,options=opts)
输出为:

from rdkit import Chem
for atom in mol.GetAtoms():
atom.SetProp('atomLabel',str(atom.GetIdx()))
mol
输出为:
7.3 多个分子按照grid显示
from rdkit.Chem import Draw
from rdkit import Chem
smis=[
'COC1=C(C=CC(=C1)NS(=O)(=O)C)C2=CN=CN3C2=CC=C3',
'C1=CC2=C(C(=C1)C3=CN=CN4C3=CC=C4)ON=C2C5=CC=C(C=C5)F',
'COC(=O)C1=CC2=CC=CN2C=N1',
'C1=C2C=C(N=CN2C(=C1)Cl)C(=O)O',
]
mols=[]
for smi in smis:
mol = Chem.MolFromSmiles(smi)
mols.append(mol)
img=Draw.MolsToGridImage(mols,molsPerRow=4,subImgSize=(200,200),legends=['' for x in mols])
img
输出为:
7.4 多个分子基于公共骨架按照grid显示
from rdkit.Chem import Draw
from rdkit import Chem
smis=[
'COC1=C(C=CC(=C1)NS(=O)(=O)C)C2=CN=CN3C2=CC=C3',
# 'CCN(CC1=C(C=CC(=C1)C(F)(F)F)C2=CC(=C3N2C=NC=C3)CC(=O)O)C(=O)C4CC4',
'C1=CC2=C(C(=C1)C3=CN=CN4C3=CC=C4)ON=C2C5=CC=C(C=C5)F',
'COC(=O)C1=CC2=CC=CN2C=N1',
'C1=C2C=C(N=CN2C(=C1)Cl)C(=O)O',
]
template = Chem.MolFromSmiles('c1nccc2n1ccc2')
AllChem.Compute2DCoords(template)
mols=[]
for smi in smis:
mol = Chem.MolFromSmiles(smi)
AllChem.GenerateDepictionMatching2DStructure(mol,template)
mols.append(mol)
img=Draw.MolsToGridImage(mols,molsPerRow=4,subImgSize=(200,200),legends=['' for x in mols])
img

八、子结构搜索
8.1 判断是否有子结构m.HasSubstructMatch(patt)
m = Chem.MolFromSmiles('c1ccccc1OC')
patt = Chem.MolFromSmarts('OC')
flag =m.HasSubstructMatch(patt)
if flag:
print("molecu m contains group -OCH3")
else:
print("molecu m don't contain group -OCH3")
输出为:molecu m contains group -OCH3
8.2 获取第一个子结构对应的原子编号m.GetSubstructMatch()
m = Chem.MolFromSmiles('c1ccccc1OC')
patt = Chem.MolFromSmarts('OC')
flag =m.HasSubstructMatch(patt)
if flag:
atomids = m.GetSubstructMatch(patt)
print("matched atom id:",atomids)
else:
print("molecu m don't contain group -OCH3")
输出为:matched atom id: (6, 7)
8.3 获取对应所有子结构(m.GetSubstructMatches)对相应的原子编号
m = Chem.MolFromSmiles('c1ccc(OC)cc1OC')
patt = Chem.MolFromSmarts('OC')
flag =m.HasSubstructMatch(patt)
if flag:
atomids = m.GetSubstructMatches(patt)
print("matched atom id:",atomids)
else:
print("molecu m don't contain group -OCH3")
输出为:matched atom id: ((4, 5), (8, 9))
8.4 子结构搜索考虑手性
m = Chem.MolFromSmiles('CC[C@H](F)Cl')
print(m.HasSubstructMatch(Chem.MolFromSmiles('C[C@H](F)Cl')))
print(m.HasSubstructMatch(Chem.MolFromSmiles('C[C@@H](F)Cl')))
print(m.HasSubstructMatch(Chem.MolFromSmiles('CC(F)Cl')))
输出为:True
True
True
m = Chem.MolFromSmiles('CC[C@H](F)Cl')
a=m.HasSubstructMatch(Chem.MolFromSmiles('C[C@H](F)Cl'),useChirality=True)
b=m.HasSubstructMatch(Chem.MolFromSmiles('C[C@@H](F)Cl'),useChirality=True)
c=m.HasSubstructMatch(Chem.MolFromSmiles('CC(F)Cl'),useChirality=True)
print(a)
print(b)
print(c)
输出为:True
False
True
m2 = Chem.MolFromSmiles('CCC(F)Cl')
print(m2.HasSubstructMatch(Chem.MolFromSmiles('C[C@H](F)Cl'),useChirality=True))
输出为:False
九、化学转换
9.1 删除子结构 AllChem.DeleteSubstructs(m,patt)
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
m = Chem.MolFromSmiles('c1ccccc1OC')
patt = Chem.MolFromSmarts('OC')
m2=AllChem.DeleteSubstructs(m,patt)
mols=[m,m2]
Draw.MolsToGridImage(mols,molsPerRow=4,subImgSize=(200,200),legends=['' for x in mols])
输出为:
9.2 取代基替换 AllChem.ReplaceSubstructs(m,patt,repl)
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
m = Chem.MolFromSmiles('COc1c(Br)cccc1OC')
patt = Chem.MolFromSmarts('OC')
repsmis= ['F','Cl','Br','O']
mols=[]
mols.append(m)
for r in repsmis:
rep = Chem.MolFromSmarts(r)
res = AllChem.ReplaceSubstructs(m,patt,rep)
mols.extend(res)
smis = [ Chem.MolToSmiles(mol) for mol in mols]
mols = [Chem.MolFromSmiles(smi) for smi in smis]
Draw.MolsToGridImage(mols,molsPerRow=3,subImgSize=(200,200),legends=['' for x in mols])
输出为:
9.3 SAR分析——core可视化Chem.ReplaceSidechains(m1,core)
m1 = Chem.MolFromSmiles('BrCCc1cncnc1C(=O)O')
core = Chem.MolFromSmiles('c1cncnc1')
tmp = Chem.ReplaceSidechains(m1,core)
tmp
输出为: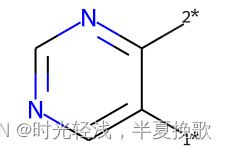
9.4 SAR分析——sidechain可视化Chem.ReplaceCore(m1,core)
m1 = Chem.MolFromSmiles('BrCCc1cncnc1C(=O)O')
core = Chem.MolFromSmiles('c1cncnc1')
tmp = Chem.ReplaceCore(m1,core)
tmp
输出为:
9.5 拆分片段 Chem.GetMolFrags(tmp,asMols=True)
9.6 对多个分子进行SAR分析(待完成)
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
m = Chem.MolFromSmiles('COc1c(Br)cccc1OC')
core = Chem.MolFromSmiles('c1c(Br)cccc1')
core_v = Chem.ReplaceSidechains(m,core)
core_v
输出为:
9.7 获取分子骨架Murcko Decomposition
from rdkit import Chem
from rdkit.Chem.Scaffolds import MurckoScaffold
m1 = Chem.MolFromSmiles('C=CC(=O)N1CCC(CC1)C2CCNC3=C(C(=NN23)C4=CC=C(C=C4)OC5=CC=CC=C5)C(=O)N')
m2 = Chem.MolFromSmiles('CCC(CC)COC(=O)C(C)NP(=O)(OCC1C(C(C(O1)(C#N)C2=CC=C3N2N=CN=C3N)O)O)OC4=CC=CC=C4')
m3= Chem.MolFromSmiles('CNC1(CCCCC1=O)C1=CC=CC=C1Cl')
core_m1 = MurckoScaffold.GetScaffoldForMol(m1)
core_m2 = MurckoScaffold.GetScaffoldForMol(m2)
core_m3 = MurckoScaffold.GetScaffoldForMol(m3)
core_mols=[core_m1,core_m2,core_m3]
Draw.MolsToGridImage(core_mols,molsPerRow=3,subImgSize=(300,300),legends=['' for x in core_mols])

9.8 最大公共子结构FindMCS
from rdkit.Chem import rdFMCS
from rdkit import Chem
mol1 = Chem.MolFromSmiles("O=C(NCc1cc(OC)c(O)cc1)CCCC/C=C/C(C)C")
mol2 = Chem.MolFromSmiles("CC(C)CCCCCC(=O)NCC1=CC(=C(C=C1)O)OC")
mol3 = Chem.MolFromSmiles("c1(C=O)cc(OC)c(O)cc1")
mols = [mol1,mol2,mol3]
res=rdFMCS.FindMCS(mols)
common=Chem.MolFromSmarts(res.smartsString)
common
输出为:
mols = [Chem.MolFromSmiles("Nc1ccccc1"*10), Chem.MolFromSmiles("Nc1ccccccccc1"*10)]
rs=rdFMCS.FindMCS(mols, timeout=1)
print(rs.canceled)
print(rs.smartsString)
输出为:True
[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:#6-[#7]-#6:[#6]:[#6]
十、指纹和相似性
10.1 拓扑指纹 Chem.RDKFingerprint(x)
ms = [Chem.MolFromSmiles('CCOC'), Chem.MolFromSmiles('CCO'), Chem.MolFromSmiles('COC')]
fps = [Chem.RDKFingerprint(x) for x in ms]
print(fps)
print(len(fps[0].ToBitString()))
print(fps[0].ToBitString())
输出为
[
2048
00000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000001000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000100000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000001000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000001000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000010000000000000000000000000000001000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000010000000000000000000000000000000000000000000100000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000010000000000000000001000000000000000000000000000000000000000000000000000000000000000000000000000000000000000
10.2 MACCS 指纹MACCSkeys.GenMACCSKeys(mol)
from rdkit.Chem import MACCSkeys
fps = [MACCSkeys.GenMACCSKeys(x) for x in ms]
print(fps)
print(len(fps[0].ToBitString()))
print(fps[0].ToBitString())
输出为
[
167
00000000000000000000000000000000000000000000000000000000000000000000000000000000000000100000010000000000000001000011000000000010000000000000000000000100010101001000100
10.3 Atom Pairs
from rdkit.Chem.AtomPairs import Pairs
ms = [Chem.MolFromSmiles('C1CCC1OCC'),Chem.MolFromSmiles('CC(C)OCC'),Chem.MolFromSmiles('CCOCC')]
pairFps = [Pairs.GetAtomPairFingerprint(x) for x in ms]
print(pairFps)
输出为
[
10.4 topological torsions
from rdkit.Chem.AtomPairs import Torsions
ms = [Chem.MolFromSmiles('C1CCC1OCC'),Chem.MolFromSmiles('CC(C)OCC'),Chem.MolFromSmiles('CCOCC')]
tts = [Torsions.GetTopologicalTorsionFingerprintAsIntVect(x) for x in ms]
print(tts)
输出为
[
10.5 摩根指纹(圆圈指纹)AllChem.GetMorganFingerprint(mol,2)
from rdkit.Chem import AllChem
m1 = Chem.MolFromSmiles('Cc1ccccc1')
fp1 = AllChem.GetMorganFingerprint(m1,2)
print(fp1)
输出为
以上几种计算指纹的方法可以转为二进制的数的RDKit拓扑指纹的长度为2048,MACCS指纹的长度为167,这么看来在内存有限的情况下,使用MACCS指纹的计算对于计算资源更友好。
10.6 基于指纹计算相似性
10.6.1 10.6.1 基于拓扑指纹 和 Tanimoto相似性方法(默认) 指纹计算3个分子的相似性
from rdkit import DataStructs
import rdkit
from rdkit import Chem
from rdkit.Chem import Draw
smis=[
'CC(=O)CC(C1=CC=C(C=C1)[N+]([O-])=O)C1=C(O)C2=CC=CC=C2OC1=O',
'CC(=O)CC(C1=CC=CC=C1)C1=C(O)C2=C(OC1=O)C=CC=C2',
'CCC(C1=CC=CC=C1)C1=C(O)C2=C(OC1=O)C=CC=C2'
]
mols =[]
for smi in smis:
m = Chem.MolFromSmiles(smi)
mols.append(m)
fps = [Chem.RDKFingerprint(x) for x in mols]
sm01=DataStructs.FingerprintSimilarity(fps[0],fps[1])
sm02=DataStructs.FingerprintSimilarity(fps[0],fps[2])
sm12=DataStructs.FingerprintSimilarity(fps[1],fps[2])
print("similarity between mol 1 and mol2: %.2f"%sm01)
print("similarity between mol 1 and mol3: %.2f"%sm02)
print("similarity between mol 2 and mol3: %.2f"%sm12)
输出为
similarity between mol 1 and mol2: 0.93
similarity between mol 1 and mol3: 0.87
similarity between mol 2 and mol3: 0.93
10.6.2 基于MACCS 指纹和Dice 相似性方法计算相似性
from rdkit import DataStructs
from rdkit.Chem import MACCSkeys
import rdkit
from rdkit import Chem
from rdkit.Chem import Draw
smis=[
'CC(=O)CC(C1=CC=C(C=C1)[N+]([O-])=O)C1=C(O)C2=CC=CC=C2OC1=O',
'CC(=O)CC(C1=CC=CC=C1)C1=C(O)C2=C(OC1=O)C=CC=C2',
'CCC(C1=CC=CC=C1)C1=C(O)C2=C(OC1=O)C=CC=C2'
]
mols =[]
for smi in smis:
m = Chem.MolFromSmiles(smi)
mols.append(m)
fps = [MACCSkeys.GenMACCSKeys(x) for x in mols]
sm01=DataStructs.FingerprintSimilarity(fps[0],fps[1],metric=DataStructs.DiceSimilarity)
sm02=DataStructs.FingerprintSimilarity(fps[0],fps[2],metric=DataStructs.DiceSimilarity)
sm12=DataStructs.FingerprintSimilarity(fps[1],fps[2],metric=DataStructs.DiceSimilarity)
print("similarity between mol 1 and mol2: %.2f"%sm01)
print("similarity between mol 1 and mol3: %.2f"%sm02)
print("similarity between mol 2 and mol3: %.2f"%sm12)
输出为
similarity between mol 1 and mol2: 0.78
similarity between mol 1 and mol3: 0.70
similarity between mol 2 and mol3: 0.92
由此可见两种计算分子指纹和相似性方法对于同样的三个分子两两之间的相似性进行比较的时候,最终结果的排序也不一致,表明相似性的比较实惠手选择方法的显著影响的。
10.7 摩根指纹的形式
from rdkit.Chem import AllChem
m1 = Chem.MolFromSmiles('Cc1ccccc1')
fp1_count = AllChem.GetMorganFingerprint(m1,2)
fp1_bit = AllChem.GetMorganFingerprintAsBitVect(m1,2,nBits=1024)
print(type(fp1_count))
print(type(fp1_bit))
输出为
10.8 摩根指纹->ECFP4 和 摩根指纹->FCFP4的比较
m1 = Chem.MolFromSmiles('c1ccccn1')
ecfp4_mg = AllChem.GetMorganFingerprint(m1,2)
fcfp4_mg = AllChem.GetMorganFingerprint(m1,2,useFeatures=True)
m1 = Chem.MolFromSmiles('c1ccccn1')
m2 = Chem.MolFromSmiles('c1ccco1')
ecfp4_m1 = AllChem.GetMorganFingerprint(m1,2)
ecfp4_m2 = AllChem.GetMorganFingerprint(m2,2)
fcfp4_m1 = AllChem.GetMorganFingerprint(m1,2,useFeatures=True)
fcfp4_m2 = AllChem.GetMorganFingerprint(m2,2,useFeatures=True)
simi_ecfp4= DataStructs.DiceSimilarity(ecfp4_m1,ecfp4_m2)
simi_fcfp4 =DataStructs.DiceSimilarity(fcfp4_m1,fcfp4_m2)
print("the similarity of the two molecules calculated by ECFP4: %.2f"%simi_ecfp4)
print("the similarity of the two molecules calculated by FCFP4: %.2f"%simi_fcfp4)
输出为
the similarity of the two molecules calculated by ECFP4: 0.36
the similarity of the two molecules calculated by FCFP4: 0.91
10.9 用户也可以自己定义atom invariants
m1 = Chem.MolFromSmiles('Cc1ccccc1')
m2 = Chem.MolFromSmiles('Cc1ncncn1')
m3 = Chem.MolFromSmiles('CC1CCCCC1')
fp1 = AllChem.GetMorganFingerprint(m1,2,invariants=[1]*m1.GetNumAtoms())
fp2 = AllChem.GetMorganFingerprint(m2,2,invariants=[1]*m2.GetNumAtoms())
fp3 =AllChem.GetMorganFingerprint(m3,2,invariants=[1]*m3.GetNumAtoms())
if(fp1==fp2):
print("If set atom invariants are the same, the fp of moleclue 1 and 2 are the same too")
if(fp1!=fp3):
print("The fp of moleclue 1 and 3 are different because the bond order will be consided in the calculation of fp ")
fp1 = AllChem.GetMorganFingerprint(m1,2,invariants=[1]*m1.GetNumAtoms(),useBondTypes=False)
fp3 = AllChem.GetMorganFingerprint(m3,2,invariants=[1]*m3.GetNumAtoms(),useBondTypes=False)
if(fp1==fp3):
print("when atom invariants are the same and bond type not considered in the calculation of fp, the fp mol 1 and 3 are the same")
输出为
If set atom invariants are the same, the fp of moleclue 1 and 2 are the same too
The fp of moleclue 1 and 3 are different because the bond order will be consided in the calculation of fp
when atom invariants are the same and bond type not considered in the calculation of fp, the fp mol 1 and 3 are the same
10.10 解释摩根指纹中bit的含义
help(AllChem.GetMorganFingerprintAsBitVect)
输出为
Help on built-in function GetMorganFingerprintAsBitVect in module rdkit.Chem.rdMolDescriptors:
GetMorganFingerprintAsBitVect(…)
GetMorganFingerprintAsBitVect( (Mol)mol, (int)radius [, (int)nBits=2048 [, (AtomPairsParameters)invariants=[] [, (AtomPairsParameters)fromAtoms=[] [, (bool)useChirality=False [, (bool)useBondTypes=True [, (bool)useFeatures=False [, (AtomPairsParameters)bitInfo=None [, (bool)includeRedundantEnvironments=False]]]]]]]]) -> ExplicitBitVect :
Returns a Morgan fingerprint for a molecule as a bit vector
C++ signature :
class ExplicitBitVect * __ptr64 GetMorganFingerprintAsBitVect(class RDKit::ROMol,unsigned int [,unsigned int=2048 [,class boost::python::api::object=[] [,class boost::python::api::object=[] [,bool=False [,bool=True [,bool=False [,class boost::python::api::object=None [,bool=False]]]]]]]])
m = Chem.MolFromSmiles('c1cccnc1C')
info={}
fp = AllChem.GetMorganFingerprint(m,2,bitInfo=info)
print("num of non-zero bit ",len(fp.GetNonzeroElements()))
print("num of keys of info",len(info.keys()))
输出为
num of non-zero bit 16
num of keys of info 16
nbitss=[64,128,256,2048]
for nbit in nbitss:
mol = Chem.MolFromSmiles('c1cccnc1C')
bi = {}
fp = AllChem.GetMorganFingerprintAsBitVect(mol, radius=2, nBits=nbit,bitInfo=bi)
print("num non zero bit in nBit=%d: %d"%(nbit,len(bi.keys())))
输出为
num non zero bit in nBit=64: 13
num non zero bit in nBit=128: 15
num non zero bit in nBit=256: 16
num non zero bit in nBit=2048: 16
nbitss=[256,2048]
bis=[]
for nbit in nbitss:
mol = Chem.MolFromSmiles('c1cccnc1C')
bi = {}
fp = AllChem.GetMorganFingerprintAsBitVect(mol, radius=2, nBits=nbit,bitInfo=bi)
bis.append(bi)
a=bis[0].values()
b=bis[1].values()
a=list(a)
b=list(b)
ab=a+b
if len(set(ab))==len(a):
print("fp info calculated by nBits=256 and 2048 are the same")
输出为
fp info calculated by nBits=256 and 2048 are the same
mol = Chem.MolFromSmiles('c1cccnc1C')
bi = {}
fp = AllChem.GetMorganFingerprintAsBitVect(mol, radius=2, nBits=256,bitInfo=bi)
for v in bi.values():
print(v)
输出为
((5, 2),)
((6, 0),)
((1, 1), (2, 1))
((3, 1),)
((0, 0), (1, 0), (2, 0), (3, 0))
((5, 0),)
((4, 2),)
((4, 0),)
((2, 2),)
((4, 1),)
((1, 2),)
((0, 2),)
((6, 1),)
((3, 2),)
((5, 1),)
((0, 1),)
10.11 获取指纹对应的结构
m = Chem.MolFromSmiles('c1cccnc1C')
env = Chem.FindAtomEnvironmentOfRadiusN(m,2,5)
amap={}
submol25=Chem.PathToSubmol(m,env,atomMap=amap)
env = Chem.FindAtomEnvironmentOfRadiusN(m,1,1)
amap={}
submol11=Chem.PathToSubmol(m,env,atomMap=amap)
env = Chem.FindAtomEnvironmentOfRadiusN(m,1,2)
amap={}
submol12=Chem.PathToSubmol(m,env,atomMap=amap)
mols=[submol25,submol11,submol12]
Draw.MolsToGridImage(mols,molsPerRow=3,subImgSize=(300,200),legends=['' for x in mols])
输出为

10.12 可视化指纹中的bit
rdkit.Chem.Draw.DrawMorganBit() 对摩根指纹中的bit 进行可视化。
rdkit.Chem.Draw.DrawRDKitBit() 对拓扑指纹中的bit 进行可视化。
10.13 可视化摩根指纹中的bit
from rdkit.Chem import Draw
from rdkit import Chem
mol = Chem.MolFromSmiles('c1cccnc1C')
bi = {}
fp = AllChem.GetMorganFingerprintAsBitVect(mol,nBits=256, radius=2, bitInfo=bi)
print(bi)
输出为
{19: ((5, 2),), 33: ((6, 0),), 64: ((1, 1), (2, 1)), 74: ((3, 1),), 81: ((0, 0), (1, 0), (2, 0), (3, 0)), 100: ((5, 0),), 121: ((4, 2),), 122: ((4, 0),), 131: ((2, 2),), 172: ((4, 1),), 175: ((1, 2),), 179: ((0, 2),), 186: ((6, 1),), 192: ((3, 2),), 195: ((5, 1),), 214: ((0, 1),)}
import matplotlib.pyplot as plt
bits =[19,64,81]
imgs=[]
for bit in bits:
mfp2_svg = Draw.DrawMorganBit(mol, bit, bi)
imgs.append(mfp2_svg)
def displayimgsinrow(imgs,col=4):
plt.figure(figsize=(20,10))
columns = col
for i, image in enumerate(imgs):
ax=plt.subplot(len(imgs) / columns + 1, columns, i + 1)
ax.set_axis_off()
plt.imshow(image)
displayimgsinrow(imgs)
输出为

蓝色: 蓝色标注说明该原子是中心原子
黄色:说明该原子是芳香原子
灰色: 说明该原子时脂肪烃原子
10.14 可视化拓扑指纹中的bit
from rdkit.Chem import Draw
from rdkit import Chem
mol = Chem.MolFromSmiles('c1cccnc1C')
rdkbi = {}
rdkfp = Chem.RDKFingerprint(mol, maxPath=2, bitInfo=rdkbi)
rdkbi
输出为
{5: [[3, 4]],
161: [[3, 4]],
294: [[0, 1], [0, 6], [1, 2]],
330: [[0, 1], [0, 6], [1, 2]],
633: [[2, 3], [4, 6]],
684: [[2, 3], [4, 6]],
744: [[5, 6]],
808: [[4, 5]],
842: [[0], [1], [2], [6]],
930: [[5]],
1026: [[0], [1], [2], [6]],
1027: [[3], [4]],
1060: [[3], [4], [5]],
1649: [[4, 5]],
1909: [[5, 6]]}
bits =[5,161,294]
imgs=[]
for bit in bits:
mfp2_svg = Draw.DrawRDKitBit(mol, bit, rdkbi)
imgs.append(mfp2_svg)
def displayimgsinrow(imgs,col=4):
plt.figure(figsize=(20,10))
columns = col
for i, image in enumerate(imgs):
ax=plt.subplot(len(imgs) / columns + 1, columns, i + 1)
ax.set_axis_off()
plt.imshow(image)
displayimgsinrow(imgs)
输出为

10.15 基于分子指纹挑选差异较大的分子
药物虚拟筛选中关键步骤挑选分子,比如筛选获得前1000个分子, 由于成本、时间等因素你想挑选100个分子进行活性测试, 如果你直接挑选前100个分子进行测试,命中率可能会降低。 一般流程是对1000个分子进行聚类,然后每一类里面挑选一个分子(或者中心分子), 这样可以提高分子骨架的多样性,从而提供虚拟筛选的成功率。
from rdkit import Chem
from rdkit.Chem.rdMolDescriptors import GetMorganFingerprint
from rdkit import DataStructs
from rdkit.SimDivFilters.rdSimDivPickers import MaxMinPicker
ms = [x for x in Chem.SDMolSupplier('data/actives_5ht3.sdf')]
while ms.count(None): ms.remove(None)
fps = [GetMorganFingerprint(x,3) for x in ms]
def distij(i,j,fps=fps):
return 1-DataStructs.DiceSimilarity(fps[i],fps[j])
picker = MaxMinPicker()
pickIndices = picker.LazyPick(distij,nfps,10,seed=23)
picks = [ms[x] for x in pickIndices]
这个部分目前是会报错的
10.16 相似性地图
相似性地图可用于可视化原子对两个分子的相似性的贡献, 该方法位于 rdkit.Chem.Draw.SimilarityMaps 模块中。
该方法支持三种类型的指纹:
atom pairs 类型表现形式 normal(default)、hashed 和 bit vector(bv) topological torsions 类型表现形式normal(default)、hashed 和 bit vector(bv) Morgan fingerprints 类型表现形式 bit vector(bv,default) 和 count vector(count)
from rdkit.Chem import Draw
from rdkit.Chem.Draw import SimilarityMaps
fp = SimilarityMaps.GetAPFingerprint(mol, fpType='normal')
fp = SimilarityMaps.GetTTFingerprint(mol, fpType='normal')
fp = SimilarityMaps.GetMorganFingerprint(mol, fpType='bv')
输出为
计算目标相似性地图,最少需要3个参数:
参考分子 目标分子 指纹函数 相似性函数(默认是 Dice similarity)
from rdkit import Chem
from rdkit.Chem.Draw import SimilarityMaps
targetmol = Chem.MolFromSmiles('COc1cccc2cc(C(=O)NCCCCN3CCN(c4cccc5nccnc54)CC3)oc21')
refmol = Chem.MolFromSmiles('CCCN(CCCCN1CCN(c2ccccc2OC)CC1)Cc1ccc2ccccc2c1')
target_mol_simi_fig, maxweight = SimilarityMaps.GetSimilarityMapForFingerprint(refmol, targetmol, SimilarityMaps.GetMorganFingerprint)
输出为

原子颜色越绿,对相似性的贡献越大。
计算相似性地图的时候,定义指纹计算方法中的参数 和相似性计算方法
from rdkit import DataStructs
from rdkit import Chem
from rdkit.Chem.Draw import SimilarityMaps
targetmol = Chem.MolFromSmiles('COc1cccc2cc(C(=O)NCCCCN3CCN(c4cccc5nccnc54)CC3)oc21')
refmol = Chem.MolFromSmiles('CCCN(CCCCN1CCN(c2ccccc2OC)CC1)Cc1ccc2ccccc2c1')
fig, maxweight = SimilarityMaps.GetSimilarityMapForFingerprint(refmol, mol, lambda m,idx: SimilarityMaps.GetMorganFingerprint(m, atomId=idx, radius=1, fpType='count'), metric=DataStructs.TanimotoSimilarity)
输出为

计算相似性的流程,首先计算原子的贡献,然后基于贡献值进行绘图。 因此也可以采用下述方式进行计算绘图。
from rdkit import DataStructs
from rdkit import Chem
from rdkit.Chem.Draw import SimilarityMaps
targetmol = Chem.MolFromSmiles('COc1cccc2cc(C(=O)NCCCCN3CCN(c4cccc5nccnc54)CC3)oc21')
refmol = Chem.MolFromSmiles('CCCN(CCCCN1CCN(c2ccccc2OC)CC1)Cc1ccc2ccccc2c1')
weights = SimilarityMaps.GetAtomicWeightsForFingerprint(refmol, mol, SimilarityMaps.GetMorganFingerprint)
fig = SimilarityMaps.GetSimilarityMapFromWeights(mol, weights)
输出为

十一、分子性质(描述符)
11.1 性质描述符计算
分子性质也被称为描述符。 RDKit中内置了大量的分子描述符的计算方法, 这些方法主要位于rdkit.Chem.Descriptors https://www.rdkit.org/docs/source/rdkit.Chem.Descriptors.html#module-rdkit.Chem.Descriptors_ 也有些常用的性质在AllChem模块下面。
RDKit 可以计算多种分子性质,如:
计算分子的The topological polar surface area (TPSA) descriptor 、logP、电荷等性质
from rdkit.Chem import Descriptors
m = Chem.MolFromSmiles('c1ccccc1C(=O)O')
tpsa_m=Descriptors.TPSA(m)
logp_m=Descriptors.MolLogP(m)
AllChem.ComputeGasteigerCharges(m)
charge_atm0=float(m.GetAtomWithIdx(0).GetProp('_GasteigerCharge'))
print("the TPSA of m is",tpsa_m)
print("the logP of m is",logp_m)
print("the gasteigerCharge of the first atom",charge_atm0)
输出为
the TPSA of m is 37.3
the logP of m is 1.3848
the gasteigerCharge of the first atom -0.04769375004654255
11.2 计算分子的价电子数目
价电子是原子在参与化学反应时能够用于成键的电子,是原子核外跟元素化合价有关的电子。
import rdkit
from rdkit.Chem.Descriptors import NumValenceElectrons
from rdkit.Chem import Descriptors
mol = Chem.MolFromSmiles("N#N")
print("the Number ValenceElectrons of mol N2 is", Descriptors.NumValenceElectrons(mol))
mol=Chem.MolFromSmiles('C(=O)=O')
print("the Number ValenceElectrons of mol CO2 is", Descriptors.NumValenceElectrons(mol))
输出为
the Number ValenceElectrons of mol N2 is 10
the Number ValenceElectrons of mol CO2 is 16
11.3 原子对性质的贡献可视化
相似性地图也可用用于性质的可视化,只要性质可以分解到原子上就可以进行可视化。
Gasteiger partial 电荷的可视化距离,自定义颜色主题jet。
from rdkit.Chem.Draw import SimilarityMaps
mol = Chem.MolFromSmiles('COc1cccc2cc(C(=O)NCCCCN3CCN(c4cccc5nccnc54)CC3)oc21')
AllChem.ComputeGasteigerCharges(mol)
contribs = [float(mol.GetAtomWithIdx(i).GetProp('_GasteigerCharge')) for i in range(mol.GetNumAtoms())]
fig = SimilarityMaps.GetSimilarityMapFromWeights(mol, contribs, colorMap='jet', contourLines=10)
输出为:

from rdkit.Chem import rdMolDescriptors
mol = Chem.MolFromSmiles('COc1cccc2cc(C(=O)NCCCCN3CCN(c4cccc5nccnc54)CC3)oc21')
contribs = rdMolDescriptors._CalcCrippenContribs(mol)
fig = SimilarityMaps.GetSimilarityMapFromWeights(mol,[x for x,y in contribs], colorMap='jet', contourLines=10)
输出为:

这里主要展示的是RDKit计算得分子性质,包括单不限于拓扑极性溶剂可及表面积、logP、部分电荷和分子的价电子数目,以及分子中各个原子对于整个分子的见电子的贡献。
总结
希望自己每天都能保持写博客的好习惯,把自己的代码笔记就极在这里,RDkit一块啃了很久的骨头,今天终于要下定决心,好好总结一波了,希望保持这个更新的频率,每天都能有所学。