【生物信息】ESTIMATE 分析免疫评分和肿瘤纯度
ESTIMATE 分析免疫评分和肿瘤纯度
背景:
从病人身上获取的肿瘤组织不仅仅包括包括肿瘤细胞,还包括与肿瘤微环境相关的很多其他细胞,比如正常的基质细胞,免疫细胞、血管内皮和血管内的血细胞。
其中比较重要的就是免疫细胞,免疫细胞的种类很多,不同类型的免疫细胞在抗肿瘤和肿瘤免疫逃逸过程中又发挥了不同的作用,肿瘤的生长、侵袭和转移与肿瘤的生长、侵袭和转移,无不与免疫细胞相关。其次就是基质细胞,基质细胞也被认为在肿瘤生长、疾病进展和耐药性中起重要作用。
所以,有人就萌生了一个想法,如果能够把肿瘤组织中与肿瘤微环境相关的免疫细胞、基质细胞和肿瘤细胞的比例或者丰度计算出来,那是不是很有意义呢?
于是来自MD Anderson cancer center的开发者就提出了一种新的算法,利用癌症样本转录谱的来推断肿瘤细胞的含量,以及浸润的免疫细胞和基质细胞,称为Estimation of STromal and Immune cells in MAlignant Tumour tissues using Expression data,简称 ESTIMATE 。
 提出 ESTIMATE 算法的文章
提出 ESTIMATE 算法的文章
Yoshihara, Kosuke et al. “Inferring tumour purity and stromal and immune cell admixture from expression data.” Nature communications vol. 4 (2013): 2612. doi:10.1038/ncomms3612
在Pubmed简单搜索,就有 3000+ 的记录,可见该方法很值得学习
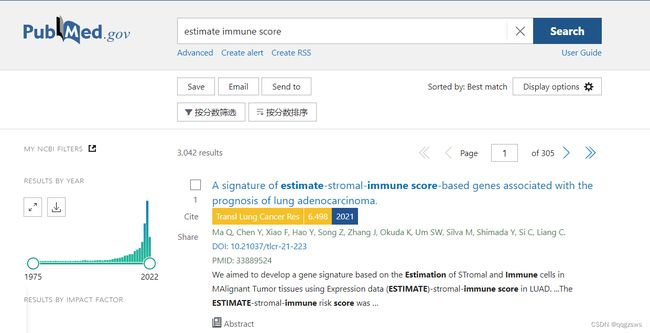 截止2022年5月Pubmed中关于ESTIMATE immune score 的检索信息,共计3042个结果
截止2022年5月Pubmed中关于ESTIMATE immune score 的检索信息,共计3042个结果
与上述CIBERSORT和ssGSEA不同的是:(1)除了免疫细胞,还能分析肿瘤细胞纯度和基质细胞的丰度;(2)关于免疫细胞,仅能计算一个总的免疫细胞评分,而无法给出每种免疫细胞的具体比例。其分析方法其实和ssGSEA有些类似,具体分析流程如下
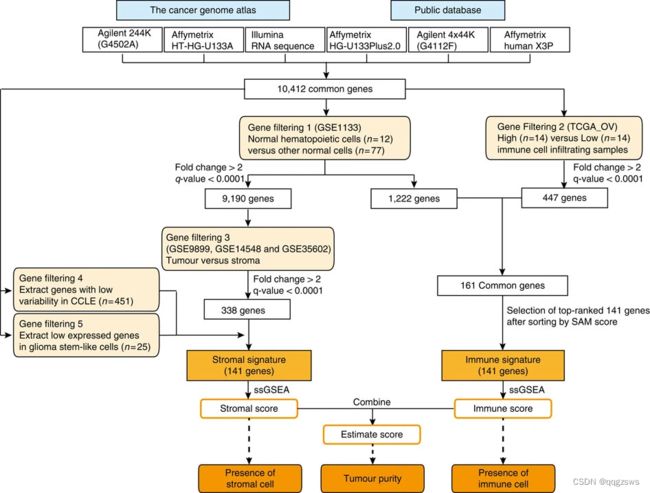
简单总结一下,作者从TCGA等公共数据库中筛选了不同平台的数据,包括Agilent, Affymetirx, Illumina RNASeq等平台,从中筛选出两个 signature,一个是Stromal signature(基质细胞特征基因),另一个是Immune signature(免疫细胞特征基因)。两个标签分别有141个基因。然后通过ssGSEA分别计算基质得分和免疫得分。最后联合这两个得分生成一个ESTIMATE score,并用于分析肿瘤纯度。该文章在2013年发表于Nature Communication杂志,
ESTIMATE官网上面有很多关于该软件的介绍,其中还对TCGA数据中的常见肿瘤进行了泛癌种的免疫浸润分析,感兴趣的同学可以访问ESTIMATE官方主页进行深入探索:
-官网链接: ESTIMATE: Home https://bioinformatics.mdanderson.org/estimate/index.html
https://bioinformatics.mdanderson.org/estimate/index.html
案例:
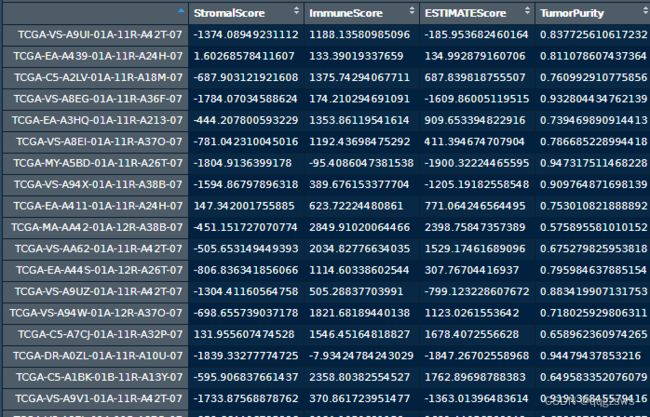
计算肿瘤样本的免疫评分,这里用的是TCGA下载的FPKM格式的表达矩阵
estimate 包计算的不止免疫评分,还包括基质评分,estimate评分及肿瘤纯度
第一步:准备R包
###计算肿瘤样本免疫评分###
#清空环境变量
rm(list=ls())
options(stringsAsFactors = F)
#安装estimate包
install.packages("estimate", repos="http://r-forge.r-project.org", dependencies=TRUE)
#加载需要用的包
library(utils)
library(estimate)
第二步:准备表达矩阵,根据数据需要考虑是否进行Log转换
##读取表达文件
load("CESC_FPKM_tumor.Rda")#加载表达矩阵文件
expr <- log2(CESC_FPKM_tumor_final + 1) #将表达量进行log转换
write.table(expr, "FPKM_tumor_log2.txt",sep = "\t",
row.names = T,col.names = NA,quote = F)第三步:将表达值矩阵先转换为gct格式
##转换GCT表达谱格式
filterCommonGenes(input.f = "FPKM_tumor_log2.txt", #输入文件名
output.f = "FPKM_tumor_log2.gct", #输出文件名
id = "GeneSymbol") #行名为gene symbol
这一步会过滤掉没有信息的基因,如下:230个基因未匹配
[1] "Merged dataset includes 10182 genes (230 mismatched)."第四步:进行estimate分析
estimateScore("FPKM_tumor_log2.gct",
"FPKM_tumor_estimate_score.txt",
platform="affymetrix") #默认平台这里可以通过下方代码查看点图,但没有很大必要
plotPurity(scores="FPKM_tumor_estimate_score.txt")
第五步:进行格式转换,整理成适合分析的格式
est <- read.table("FPKM_tumor_estimate_score.txt",
sep = "\t",row.names = 1,check.names = F,stringsAsFactors = F,header = T)
est <- est[,-1] #移除第一列
colnames(est) <- est[1,] #设置列名
est <- as.data.frame(t(est[-1,]))
rownames(est) <- colnames(expr)
write.table(est, file = "FPKM_tumor_estimate_score.txt",
sep = "\t",row.names = T,col.names = NA,quote = F)最终得到如下表所示结果