识别差异表达蛋白编码基因
识别差异表达蛋白编码基因
- 题目回顾
- 1.识别差异表达蛋白编码基因
-
- 1.1文件读入
- 1.2 预处理
- 1.3 t检验
- 1.4 fold change
- 1.5合并
- 2.绘制火山图
-
- 2.1最简单的火山图绘制:
- 2.2关于ggplot2的其他细节用法
- 3.绘制热图
-
- 3.1 选出up、down基因各100个
- 3.2 作图
此文灵感来自 一个师姐的文章
题目回顾
1.利用T检验结合FC的方法识别差异表达蛋白编码基因及lncRNA(ESC与CM之间的差异)注:考虑数据的预处理(例如 删除全部样本低表达基因,log转化)。(结果文件,基因ID ESC均值 CM均值 FC P值 FDR值)
2.绘制差异表达蛋白编码基因及lncRNA火山图(ggplot2)
3.利用差异表达mRNA的表达谱进行聚类分析(pheatmap)
1.识别差异表达蛋白编码基因
利用T检验结合FC的方法识别差异表达蛋白编码基因及lncRNA(ESC与CM之间的差异)注:考虑数据的预处理(例如 删除全部样本低表达基因,log转化)。
结果文件:基因ID ESC均值 CM均值 FC P值 FDR值
1.1文件读入
code 1.1
##---读入文件---##
setwd("D:/QQ/1903786521/FileRecv")
pro.count <- read.table("pro_counts.txt",header = TRUE,row.names = 1,stringsAsFactors = F)
#> View(pro.count)
pro.fpkm <- read.table("protein_coding.fpkm.landscape.txt",header = TRUE,row.names = 1,stringsAsFactors = F)
#> View(pro.fpkm)
这里的header = TRUE可以理解为文件有列名
row.names = 1可以理解为第一列是行名,如果没有这句,后续处理依据FPKM值筛选基因时要考虑第一列不是数值而是字符1。
更多关于read.table()的内容
1.2 预处理
如果不加row.names=1,一种解决方式是把除第一列外的dataframe搬到一个矩阵(这里叫pro.mtx)里,然后在矩阵里预处理,见1.2.a。
这样会丢掉ID,最后找ID比较麻烦,巨不推荐 这是一只脑残喵的最初做法
code 1.2.a
##---预处理---##
pro.mtx <- pro.fpkm[,2:length(pro.fpkm[1,])]
pro.mtx <- as.matrix(pro.mtx)
sum = vector() #也可sum = NULL
for(i in 1:nrow(pro.mtx)){
if(all(pro.mtx[i,]>1)){
sum <- c(sum,i)
}
}
#> nrow(pro.mtx)
#[1] 15574
#> length(sum)
#[1] 7092
#去掉fpkm值小于1的基因之后,从15574个基因变为7092个基因
pro.rest <- pro.mtx[sum,]
pro.log <- log2(pro.rest+0.05)
如果行名列名都正常存在,预处理如:1.2.b
code 1.2.b
##---预处理---##
pro.rest <- data.frame()
for(i in 1:nrow(pro.fpkm)){
if(all(pro.fpkm[i,]>1)){
pro.rest <- rbind(pro.rest,pro.fpkm[i,])
}
}
pro.log <- log2(pro.rest+0.05)
为什么筛除FPKM<=1的基因?
因为取后面要取log2,如果不筛出来的话,会使取过log2的数出现小于0的情况。
同时,依题意:“删除全部样本低表达基因”
为什么要取log2?
依稀记得好像可以“使数据更符合正态分布”
why? 未知&&搜索ing~
1.3 t检验
t检验见1.3
code 1.3
##---t检验---##
pvalues <- vector()
for(i in 1:nrow(pro.log)){
pvalue <- t.test(pro.log[i,grep("ESC",colnames(pro.log))],
pro.log[i,grep("CM",colnames(pro.log))])$p.value
pvalues <- c(pvalues,pvalue)
}
fdr <- p.adjust(pvalues)
grep("ESC",colnames(pro.log))的作用是:把esc组的列找出来(文件的列名为ESC1、ESC2、ESC3…)
1.4 fold change
code 1.4
##---fold change---##
mean_esc <- apply(pro.rest[,grep("ESC",colnames(pro.rest))], 1, mean)
mean_cm <- apply(pro.rest[,grep("CM",colnames(pro.rest))], 1, mean)
fc <- mean_cm / mean_esc
fc.log <- log2(fc)
1.5合并
合并的时候,依据题意,第一列是ID,刚刚把ID丢掉的可怜孩纸,现在需要把ID找回来了,如1.5.a
code 1.5.a
##---合并---##
id <- vector()
for(i in 1:length(sum)){
id[i] <- as.character(pro.fpkm[sum[i],1])
}
result <- data.frame(id=id,esc=mean_esc,cm=mean_cm,log2fc=fc.log,pvalues=pvalues,fdr=fdr)
write.csv(result,"result.csv")
行名列名都正常的话,如1.5.b
code 1.5.b
result <- data.frame(esc=mean_esc,cm=mean_cm,log2fc=fc.log,pvalues=pvalues,fdr=fdr)
write.csv(result,"result.csv")
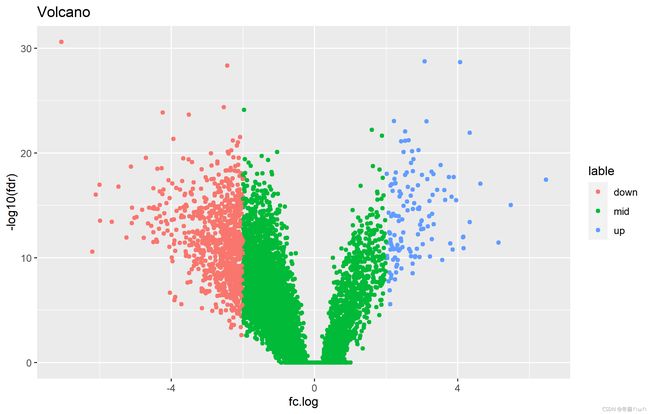
2.绘制火山图
2.1最简单的火山图绘制:
##---volcano---##
library(ggplot2)
result$lable <- ifelse(result$pvalues>0.01,"mid",
ifelse(result$log2fc > 2,"up",
ifelse(result$log2fc < -2,"down","mid")))
ggplot(result, aes(x=fc.log, y=-log10(fdr)))+geom_point(aes(color=lable))+
labs(title="Volcano")
ggsave("volcano.png")
这里加一个标签区分上调下调基因,注意">"、"<"符号养成加空格的习惯,否则写成<-2容易变成赋值句
某只傻喵在这里摔了n次终于爬起来了
如图:
2.2关于ggplot2的其他细节用法
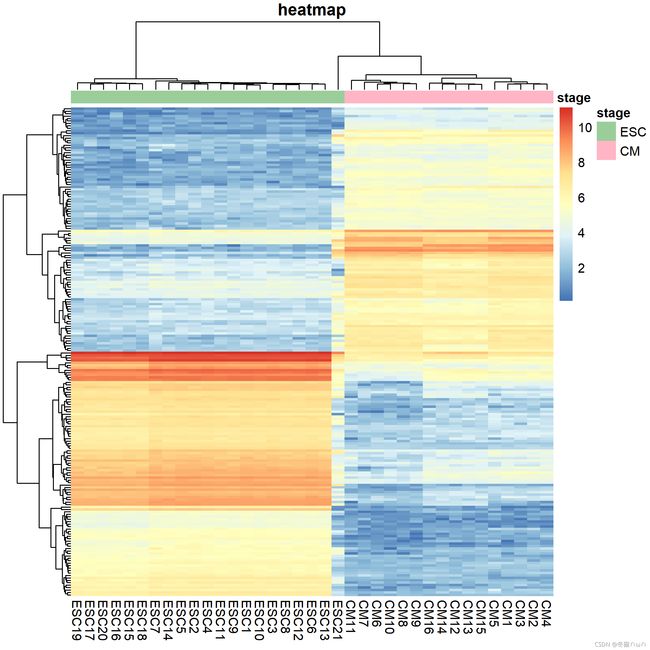
3.绘制热图
3.1 选出up、down基因各100个
多一些或者少一些应该都可吧
code 3.1
##---heatmap---##
result.updown <- result[result$lable!="mid",]
result.updown <- result.updown[order(-result.updown$log2fc),]
pro.up.name <- rownames(head(result.updown,100))
pro.down.name <- rownames(tail(result.updown,100))
pro.sig <- pro.log[c(pro.up.name,pro.down.name),]
其实pro.sig <- pro.log[c(pro.up.name,pro.down.name),]这里似乎应该是用pro.sig <- pro.fpkm[c(pro.up.name,pro.down.name),],但是作图效果不理想(蓝色糊一大片),于是自作主张改成了log2后的值。
3.2 作图
code 3.2
sample.class <- data.frame(stage = factor(c(rep("ESC", 21), rep("CM", 16))))
rownames(sample.class) <- colnames(pro.sig)
sample.color <- list(stage = c(ESC = "#9BCD9B", CM = "#FFB5C5"))
pro.sig <- as.matrix(pro.sig)
pheatmap(pro.sig,annotation_col=sample.class,annotation_colors=sample.color,
show_colnames=T,show_rownames=F,main="heatmap",filename="heatmap2.png")
其他的,持续更新ing~
比如:取出某基因的一整行,问:你们都大于1吗?这时排在第一位的(基因名)发话了:“我压根不是数字,你竟然问我是不是大于1?”所以最终的反馈都会是FALSE,因此,不容易直接筛选出想要的基因。 ↩︎