- 509. 斐波那契数(每日一题)
lzyprime
lzyprime博客(github)创建时间:2021.01.04qq及邮箱:2383518170leetcode笔记题目描述斐波那契数,通常用F(n)表示,形成的序列称为斐波那契数列。该数列由0和1开始,后面的每一项数字都是前面两项数字的和。也就是:F(0)=0,F(1)=1F(n)=F(n-1)+F(n-2),其中n>1给你n,请计算F(n)。示例1:输入:2输出:1解释:F(2)=F(1)+
- 数组去重
好奇的猫猫猫
整理自js中基础数据结构数组去重问题思考?如何去除数组中重复的项例如数组:[1,3,4,3,5]我们在做去重的时候,一开始想到的肯定是,逐个比较,外面一层循环,内层后一个与前一个一比较,如果是久不将当前这一项放进新的数组,挨个比较完之后返回一个新的去过重复的数组不好的实践方式上述方法效率极低,代码量还多,思考?有没有更好的方法这时候不禁一想当然有了!!!hashtable啊,通过对象的hash办法
- 【讲解】怎么消除妊娠纹
poyan7160
女人是脆弱的,尤其是孕期的女性。辛辛苦苦怀胎十月,经历一次深到骨子里的痛还不够,无奈还要留下一身的妊娠纹。母亲是伟大的,但也是要付出代价的,妊娠纹就是最好的证明。可是,难道真的要带着妊娠纹过一辈子吗?不,坚决不!接下来新时代辣妈告诉你怎么去除妊娠纹?怎么去除妊娠纹——根据肌肤需要补充水分就像敷面膜那样,大家都知道敷面膜的目的是为了给肌肤补充水分。水分对一个人的肌肤很重要,只有有了足够的水分,肌肤才
- 一文掌握python面向对象魔术方法(二)
程序员neil
pythonpython开发语言
接上篇:一文掌握python面向对象魔术方法(一)-CSDN博客目录六、迭代和序列化:1、__iter__(self):定义迭代器,使得类可以被for循环迭代。2、__getitem__(self,key):定义索引操作,如obj[key]。3、__setitem__(self,key,value):定义赋值操作,如obj[key]=value。4、__delitem__(self,key):定义
- 七月你好
茗蕙原创
告别了说变天就变的六月正值七月酷暑之时没有嬉戏的鱼水之乐站在窗边抬头望着蔚蓝天空万里无云万里天七月你好在月末的几天里在家期盼出门时的喜悦别样的天气别样的心情七月你好让大地经受着煎熬让空气中充呲着滚滚热浪去抵御往年严冬带来的湿气七月你好你的到来如逢甘露愿你带来的温暖去除病菌让人们重新看到生活的希望向往南山一角
- 一分钟学会刷牙,受用终生!
好易康
讲真,刷了十几二十年牙,没刷对过一次......来来来,划重点,更重要的是执行:①每天刷牙2次,②每次刷牙2~3分钟,③每3个月更换牙刷。最后,请使用正确的刷牙方法:巴氏(BASS)刷牙法undefined_腾讯视频视频来源ADA美国牙医协会巴氏刷牙法又称龈沟清扫法或水平颤动法。是由美国牙科协会推荐的一种有效去除龈缘附近及龈沟内菌斑的方法。刷牙不仅是刷牙齿,同时也要刷牙龈。因为口腔与细菌的战场就在
- 2024春节微信红包封面序列号大全一览
帮忙赚赏金
2024微信红包封面序列号哪里领取红包封面领取微信搜索公众号:【艺间封面】千万红包封面等你领取2024微信红包封面免费序列号如何设置微信红包封面?1.打开微信,点击好友选择红包。2.单击红包封面。3.单击“添加红包封面”。4.输入接收序列号。来一波免费的微信红包封面序列号微信红包封面序列号红包封面领取微信搜索公众号:艺间封面千万红包封面等你领取微信红包封面序列号kGnkrbw5a7N微信红包封面序
- 【2021-01-24】人生十三信条
wc的一些事一些情
23:59一个人应当有良好的礼貌来突出他特有的天性。人人都喜欢出人头地,但这不应当引起别人的讨厌。——歌德1、节制:食不可过饱,饮不得过量。2、缄默:避免无谓闲扯,言谈必须对人有益。3、秩序:生活物品要放置有序,工作时间要合理安排。4、决心:要做的事就下决心去做,决心做的事一定要按时完成。5、节俭:不奢侈浪费,任何花费都要做到有益,不论是于人,还是于己。6、勤勉:珍惜每一刻时间,去除一切不必要之举
- 每日OJ_牛客_马戏团(模拟最长上升子序列)
GR鲸鱼
c++算法开发语言牛客数据结构
目录牛客_马戏团(模拟最长上升子序列)解析代码牛客_马戏团(模拟最长上升子序列)马戏团__牛客网搜狐员工小王最近利用假期在外地旅游,在某个小镇碰到一个马戏团表演,精彩的表演结束后发现团长正和大伙在帐篷前激烈讨论,小王打听了下了解到,马戏团正打算出一个新节目“最高罗汉塔”,即马戏团员叠罗汉表演。考虑到安全因素,要求叠罗汉过程中,站在某个人肩上的人应该既比自己矮又比自己瘦,或相等。团长想要本次节目中的
- 2024微信红包封面怎么领取免费的?(红包封面序列号获取方法)
帮忙赚赏金
2024微信红包封面怎么领取免费的?(红包封面序列号获取方法)在中国,微信几乎成为了人们生活中不可或缺的一部分,而微信红包更是成为了人们表达祝福和送礼的一种形式。微信红包不仅方便快捷,还能够增添节日气氛和人与人之间的情感交流。然而,有时候我们想要定制一个特殊的微信红包封面,以更好地展现自己的个性和情感,但又担心定制费用过高。那么,如何才能免费获取2024微信红包封面的序列号呢?下面将为您详细介绍一
- Python 推导式(Comprehensions)
戒灵
1,列表推导式num=[1,2,-5,10,-7,5,7,-1]filtered_and_squared=[x**2forxinnumifx>0]print(filtered_and_squared)迭代器(iterator)遍历输入序列num的每个成员x断言式判断每个成员是否大于零如果成员大于零,则被交给输出表达式,平方之后成为输出列表的成员。列表推导式被封装在一个列表中,所以很明显它能够立即生
- KVM+GFS分布式存储系统构建KVM高可用
henan程序媛
分布式GFS高可用KVM
一、案列分析1.1案列概述本章案例主要使用之前章节所学的KVM及GlusterFs技术,结合起来从而实现KVM高可用。利用GlusterFs分布式复制卷,对KVM虚拟机文件进行分布存储和冗余。分布式复制卷主要用于需要冗余的情况下把一个文件存放在两个或两个以上的节点,当其中一个节点数据丢失或者损坏之后,KVM仍然能够通过卷组找到另一节点上存储的虚拟机文件,以保证虚拟机正常运行。当节点修复之后,Glu
- Codeforces Round 972 (Div. 2) A-C 题解
AKDreamer_HeXY
Codeforces比赛题解c++算法动态规划数据结构贪心算法
本来以为B2难度会1900什么的,结果感觉1200还没有,先做的B1,后悔了QwQ关于我现场没切出C这件事……现场排名:A.SimplePalindrome题意构造一个长度为nnn的字符串,只包含aeiou五种字母,需要使得构造出来的字符串所包含的回文子序列数量最小思路当n≤5n\le5n≤5时,只要555个字母不重复出现都是最优情况当n>5n>5n>5时,可以证明:把相同字母放在一起是最优情况:
- 算法刷题:300. 最长递增子序列、674. 最长连续递增序列、718. 最长重复子数组、1143. 最长公共子序列
哆来咪咪咪
算法
300.最长递增子序列1.dp定义:dp[i]表示i之前包括i的以nums[i]结尾的最长递增子序列的长度2.递推公式:if(nums[i]>nums[j])dp[i]=max(dp[i],dp[j]+1);注意这里不是要dp[i]与dp[j]+1进行比较,而是我们要取dp[j]+1的最大值。3.初始化:每一个i,对应的dp[i](即最长递增子序列)起始大小至少都是1.classSolution{
- python中的迭代器有什么用
hakesashou
python基础知识python开发语言
什么是Python迭代器?迭代器(Iterator):迭代器可以看作是一个特殊的对象,每次调用该对象时会返回自身的下一个元素,从实现上来看,一个迭代器对象必须是定义了__iter__()方法和next()方法的对象。1、Python的Iterator对象表示的是一个数据流,可以把这个数据流看做是一个有序序列,但我们却不能提前知道序列的长度,所以Iterator的计算是惰性的,只有在需要返回下一个数
- 如何区分Python中数据类型可变还是不可变
秸秆混凝烧结工程师
关键字改变元素值,内存地址发生改变,被称为数据内型不可变如string,元组,存储数据类型单一,不能同时存在两个数据类型,新增元素后,表容量,元素个数,元素存储区ID改变,典型的内置元素一体存储法;改变元素值,但是内存地址不改变就是可变数据内型,如list,存储元素可以不同,删除,新增,插入,表序列不改变,扩展表容量时,对象地址ID不变,属于顺序表的,分离式存储结构,外置元素法,python中不可
- 基于TRIZ的救援机器人轻量化设计
天行健王春城老师
TRIZ机器人
在救援机器人设计中,轻量化是一个至关重要的目标,它直接关系到机器人的便携性、运输效率以及在复杂环境中的作业能力。TRIZ理论为我们提供了一套系统化的工具和方法,用于解决设计过程中遇到的各种挑战,特别是在实现轻量化目标时,TRIZ能够帮助我们识别并消除设计中的冗余与低效部分,同时保留或增强其关键功能。具体如深圳天行健企业管理咨询公司下文所述:1.功能分析与矛盾识别TRIZ理论强调对系统功能的深入分析
- 机电综合管理系统架构
小熊coder
机载系统系统架构
文章目录一、机电综合管理系统架构1.系统概述2.架构层次3.核心组件二、余度管理1.余度概述2.硬件冗余3.软件冗余4.通信冗余三、总线架构1.MIL-STD-1553B总线2.ARINC429总线3.ARINC629总线4.AFDX/ARINC664总线四、未来发展趋势1.分布式架构2.高速网络3.智能化与自动化结语机电综合管理系统(ElectromechanicalManagementSyst
- 【Java】面试题31:栈的压入,弹出序列
小小核桃
剑指offerjava版
~~题目:~~输入两个整数序列,第一个序列表示栈的压入顺序,请判断第二个序列是否为该栈的弹出顺序。假设压入栈的所有数字均不相等。例如,序列{1,2,3,4,5}是某栈的压栈序列,序列{4,5,3,2,1}是该压栈序列对应的一个弹出序列,但{4,3,5,1,2}就不可能是该栈序列的弹出序列。思路:首先借助一个辅助栈,把输入的第一个序列中的数字依次压入该辅助栈,并按照第二个序列的顺序依次从该栈中弹出数
- ODOO不同版本与平台选择
chouchengyin2080
c#操作系统运维
1.10.0vs11.0vs8.0截至2017年底,最新的ODOO发布版为ODOO11.0,但功能上有一定精简(去除财务模块,去除工作流支持),技术上变动较大(代码逐步迁移至Python3,前端框架改写得抽象)。所以如果是从生产使用的角度来讲,ODOO10.0是当前最好选择,因为其更稳定,第三方模块也更多更全面。而如果是ODOO技术爱好从业者,则逐步迁移至ODOO11.0也有必要,因为其底层技术架
- SQLite的入门级项目学习记录(二)
深蓝海拓
SQLite学习笔记sqlite学习数据库
再补充一些基础知识:并行操作的问题1、可以多游标同时运行SQLite,对于同一个连接sqlite3.connect(db_file),可以同时创建多个游标,每个游标都是独立的,可以执行各自的SQL命令序列。importsqlite3#创建数据库连接conn=sqlite3.connect('example.db')#创建第一个游标cursor1=conn.cursor()cursor1.execu
- Pyorch中 nn.Conv1d 与 nn.Linear 的区别
迪三
#NN_Layer神经网络
即一维卷积层和全联接层的区别nn.Conv1d和nn.Linear都是PyTorch中的层,它们用于不同的目的,主要区别在于它们处理输入数据的方式和执行的操作类型。nn.Conv1d通过应用滑动过滤器来捕捉序列数据中的局部模式,适用于处理具有时间或序列结构的数据。nn.Linear通过将每个输入与每个输出相连接,捕捉全局关系,适用于将输入数据作为整体处理的任务。1.维度与输入nn.Conv1d(一
- 老生常谈:MySQL高可用架构
我有一头小花驴
mysql架构数据库
引言“高可用”是互联网一个永恒的话题,先避开MySQL不谈,为了保证各种服务的高可用有几种常用的解决方案。服务冗余:把服务部署多份,当某个节点不可用时,切换到其他节点。服务冗余对于无状态的服务是相对容易的。服务备份:有些服务是无法同时存在多个运行时的,比如说:Nginx的反向代理,一些集群的leader节点。这时可以存在一个备份服务,处于随时待命状态。自动切换:服务冗余之后,当某个节点不可用时,要
- Apache HBase基础(基本概述,物理架构,逻辑架构,数据管理,架构特点,HBase Shell)
May--J--Oldhu
HBaseHBaseshellhbase物理架构hbase逻辑架构hbase
NoSQL综述及ApacheHBase基础一.HBase1.HBase概述2.HBase发展历史3.HBase应用场景3.1增量数据-时间序列数据3.2信息交换-消息传递3.3内容服务-Web后端应用程序3.4HBase应用场景示例4.ApacheHBase生态圈5.HBase物理架构5.1HMaster5.2RegionServer5.3Region和Table6.HBase逻辑架构-Row7.
- 数据分析-24-时间序列预测之基于keras的VMD-LSTM和VMD-CNN-LSTM预测风速
皮皮冰燃
数据分析数据分析
文章目录1普通的LSTM模型1.1数据重采样1.2数据标准化1.3切分窗口1.4划分数据集1.5建立模型1.6预测效果2VMD-LSTM模型2.1VMD分解时间序列2.2对每一个IMF建立LSTM模型2.2.1IMF1—LSTM2.2.2IMF2-LSTM2.2.3统一代码2.3评估效果3CNN-LSTM模型3.1数据预处理3.2建立模型3.3效果预测4VMD-CNN-LSTM模型4.1VMD分解
- 车载以太网之SOME/IP
IT_码农
车载以太网车载以太网SOME/IP
整体介绍SOME/IP(全称为:Scalableservice-OrientedMiddlewarEoverIP),是运行在车载以太网协议栈基础之上的中间件,或者也可以称为应用层软件。发展历程AUTOSAR4.0-完成宝马SOME/IP消息的初步集成;AUTOSAR4.1-支持SOME/IP-SD及其发布/订阅功能;AUTOSAR4.2-添加transformer用于序列化以及其他相关优化;AUT
- 微信红包封面序列号兑换码大全免费2024最新龍年
全网优惠分享
每当月初的时候,我们都期待着的就是那一句话:“老板发红包了!”纷纷掏出手机,急切地等待着微信红包的到来。红包弹出的那一瞬间,我们的心情也跟着变得愉悦起来。这看似微不足道的小红包,却蕴含着我们对生活的期盼和希望。它不仅仅是简单的财富分享,更是一种情感的表达。微.信搜索:「封面院」关注公众号可领取红包封面序列号。最新微信红包封面序列号:先到先得,抢完为止:1、pdiqgLsY1lR2、vC8tY0VR
- 1.6编程基础之一维数组
伶俐角少儿编程
C++入门篇算法c++数据结构
文章目录01:与指定数字相同的数的个数02:陶陶摘苹果03:计算书费04:数组逆序重放05:年龄与疾病06:校门外的树07:有趣的跳跃08:石头剪刀布09:向量点积计算10:大整数加法11:大整数减法12:计算2的N次方13:大整数的因子14:求10000以内n的阶乘15:阶乘和01:与指定数字相同的数的个数总时间限制:1000ms内存限制:65536kB描述输出一个整数序列中与指定数字相同的数的
- 动态生成的html元素绑定click事件
.NET跨平台
Jquery及其组件htmljquery
第一篇博客,开启技术博客的生涯,欢迎大家批评指教(坚信妹子也可以做好程序猿)今天想说帮公司做项目的时候遇到的一个小问题,动态添加html元素以后再去事件监听出问题。在实际开发中会遇到要给动态生成的html元素绑定触发事件的情况。就是上面的一张表格要动态实现添加行,然后序列号还要随着增加,当删除的时候序列号依旧是按顺序排列。刚开始使用jQuery的on方法来解决,但是发现一个问题会出现事件绑定很多次
- 通过C# 裁剪PDF页面
Eiceblue
C#.NETPDFc#pdf开发语言visualstudio
在处理PDF文档时,有时需要精确地裁剪页面以适应特定需求,比如去除广告、背景信息或者仅仅是为了简化文档内容。本文将指导如何使用免费.NET控件通过C#实现裁剪PDF页面。免费库FreeSpire.PDFfor.NET支持在.NET(C#,VB.NET,ASP.NET,.NETCore)程序中实现创建、操作、转换和打印PDF文档等操作。可以从以下链接下载产品包后手动添加引用,或者直接通过NuGet安
- 对股票分析时要注意哪些主要因素?
会飞的奇葩猪
股票 分析 云掌股吧
众所周知,对散户投资者来说,股票技术分析是应战股市的核心武器,想学好股票的技术分析一定要知道哪些是重点学习的,其实非常简单,我们只要记住三个要素:成交量、价格趋势、振荡指标。
一、成交量
大盘的成交量状态。成交量大说明市场的获利机会较多,成交量小说明市场的获利机会较少。当沪市的成交量超过150亿时是强市市场状态,运用技术找综合买点较准;
- 【Scala十八】视图界定与上下文界定
bit1129
scala
Context Bound,上下文界定,是Scala为隐式参数引入的一种语法糖,使得隐式转换的编码更加简洁。
隐式参数
首先引入一个泛型函数max,用于取a和b的最大值
def max[T](a: T, b: T) = {
if (a > b) a else b
}
因为T是未知类型,只有运行时才会代入真正的类型,因此调用a >
- C语言的分支——Object-C程序设计阅读有感
darkblue086
applec框架cocoa
自从1972年贝尔实验室Dennis Ritchie开发了C语言,C语言已经有了很多版本和实现,从Borland到microsoft还是GNU、Apple都提供了不同时代的多种选择,我们知道C语言是基于Thompson开发的B语言的,Object-C是以SmallTalk-80为基础的。和C++不同的是,Object C并不是C的超集,因为有很多特性与C是不同的。
Object-C程序设计这本书
- 去除浏览器对表单值的记忆
周凡杨
html记忆autocompleteform浏览
&n
- java的树形通讯录
g21121
java
最近用到企业通讯录,虽然以前也开发过,但是用的是jsf,拼成的树形,及其笨重和难维护。后来就想到直接生成json格式字符串,页面上也好展现。
// 首先取出每个部门的联系人
for (int i = 0; i < depList.size(); i++) {
List<Contacts> list = getContactList(depList.get(i
- Nginx安装部署
510888780
nginxlinux
Nginx ("engine x") 是一个高性能的 HTTP 和 反向代理 服务器,也是一个 IMAP/POP3/SMTP 代理服务器。 Nginx 是由 Igor Sysoev 为俄罗斯访问量第二的 Rambler.ru 站点开发的,第一个公开版本0.1.0发布于2004年10月4日。其将源代码以类BSD许可证的形式发布,因它的稳定性、丰富的功能集、示例配置文件和低系统资源
- java servelet异步处理请求
墙头上一根草
java异步返回servlet
servlet3.0以后支持异步处理请求,具体是使用AsyncContext ,包装httpservletRequest以及httpservletResponse具有异步的功能,
final AsyncContext ac = request.startAsync(request, response);
ac.s
- 我的spring学习笔记8-Spring中Bean的实例化
aijuans
Spring 3
在Spring中要实例化一个Bean有几种方法:
1、最常用的(普通方法)
<bean id="myBean" class="www.6e6.org.MyBean" />
使用这样方法,按Spring就会使用Bean的默认构造方法,也就是把没有参数的构造方法来建立Bean实例。
(有构造方法的下个文细说)
2、还
- 为Mysql创建最优的索引
annan211
mysql索引
索引对于良好的性能非常关键,尤其是当数据规模越来越大的时候,索引的对性能的影响越发重要。
索引经常会被误解甚至忽略,而且经常被糟糕的设计。
索引优化应该是对查询性能优化最有效的手段了,索引能够轻易将查询性能提高几个数量级,最优的索引会比
较好的索引性能要好2个数量级。
1 索引的类型
(1) B-Tree
不出意外,这里提到的索引都是指 B-
- 日期函数
百合不是茶
oraclesql日期函数查询
ORACLE日期时间函数大全
TO_DATE格式(以时间:2007-11-02 13:45:25为例)
Year:
yy two digits 两位年 显示值:07
yyy three digits 三位年 显示值:007
- 线程优先级
bijian1013
javathread多线程java多线程
多线程运行时需要定义线程运行的先后顺序。
线程优先级是用数字表示,数字越大线程优先级越高,取值在1到10,默认优先级为5。
实例:
package com.bijian.study;
/**
* 因为在代码段当中把线程B的优先级设置高于线程A,所以运行结果先执行线程B的run()方法后再执行线程A的run()方法
* 但在实际中,JAVA的优先级不准,强烈不建议用此方法来控制执
- 适配器模式和代理模式的区别
bijian1013
java设计模式
一.简介 适配器模式:适配器模式(英语:adapter pattern)有时候也称包装样式或者包装。将一个类的接口转接成用户所期待的。一个适配使得因接口不兼容而不能在一起工作的类工作在一起,做法是将类别自己的接口包裹在一个已存在的类中。 &nbs
- 【持久化框架MyBatis3三】MyBatis3 SQL映射配置文件
bit1129
Mybatis3
SQL映射配置文件一方面类似于Hibernate的映射配置文件,通过定义实体与关系表的列之间的对应关系。另一方面使用<select>,<insert>,<delete>,<update>元素定义增删改查的SQL语句,
这些元素包含三方面内容
1. 要执行的SQL语句
2. SQL语句的入参,比如查询条件
3. SQL语句的返回结果
- oracle大数据表复制备份个人经验
bitcarter
oracle大表备份大表数据复制
前提:
数据库仓库A(就拿oracle11g为例)中有两个用户user1和user2,现在有user1中有表ldm_table1,且表ldm_table1有数据5千万以上,ldm_table1中的数据是从其他库B(数据源)中抽取过来的,前期业务理解不够或者需求有变,数据有变动需要重新从B中抽取数据到A库表ldm_table1中。
- HTTP加速器varnish安装小记
ronin47
http varnish 加速
上午共享的那个varnish安装手册,个人看了下,有点不知所云,好吧~看来还是先安装玩玩!
苦逼公司服务器没法连外网,不能用什么wget或yum命令直接下载安装,每每看到别人博客贴出的在线安装代码时,总有一股羡慕嫉妒“恨”冒了出来。。。好吧,既然没法上外网,那只能麻烦点通过下载源码来编译安装了!
Varnish 3.0.4下载地址: http://repo.varnish-cache.org/
- java-73-输入一个字符串,输出该字符串中对称的子字符串的最大长度
bylijinnan
java
public class LongestSymmtricalLength {
/*
* Q75题目:输入一个字符串,输出该字符串中对称的子字符串的最大长度。
* 比如输入字符串“google”,由于该字符串里最长的对称子字符串是“goog”,因此输出4。
*/
public static void main(String[] args) {
Str
- 学习编程的一点感想
Cb123456
编程感想Gis
写点感想,总结一些,也顺便激励一些自己.现在就是复习阶段,也做做项目.
本专业是GIS专业,当初觉得本专业太水,靠这个会活不下去的,所以就报了培训班。学习的时候,进入状态很慢,而且当初进去的时候,已经上到Java高级阶段了,所以.....,呵呵,之后有点感觉了,不过,还是不好好写代码,还眼高手低的,有
- [能源与安全]美国与中国
comsci
能源
现在有一个局面:地球上的石油只剩下N桶,这些油只够让中国和美国这两个国家中的一个顺利过渡到宇宙时代,但是如果这两个国家为争夺这些石油而发生战争,其结果是两个国家都无法平稳过渡到宇宙时代。。。。而且在战争中,剩下的石油也会被快速消耗在战争中,结果是两败俱伤。。。
在这个大
- SEMI-JOIN执行计划突然变成HASH JOIN了 的原因分析
cwqcwqmax9
oracle
甲说:
A B两个表总数据量都很大,在百万以上。
idx1 idx2字段表示是索引字段
A B 两表上都有
col1字段表示普通字段
select xxx from A
where A.idx1 between mmm and nnn
and exists (select 1 from B where B.idx2 =
- SpringMVC-ajax返回值乱码解决方案
dashuaifu
AjaxspringMVCresponse中文乱码
SpringMVC-ajax返回值乱码解决方案
一:(自己总结,测试过可行)
ajax返回如果含有中文汉字,则使用:(如下例:)
@RequestMapping(value="/xxx.do") public @ResponseBody void getPunishReasonB
- Linux系统中查看日志的常用命令
dcj3sjt126com
OS
因为在日常的工作中,出问题的时候查看日志是每个管理员的习惯,作为初学者,为了以后的需要,我今天将下面这些查看命令共享给各位
cat
tail -f
日 志 文 件 说 明
/var/log/message 系统启动后的信息和错误日志,是Red Hat Linux中最常用的日志之一
/var/log/secure 与安全相关的日志信息
/var/log/maillog 与邮件相关的日志信
- [应用结构]应用
dcj3sjt126com
PHPyii2
应用主体
应用主体是管理 Yii 应用系统整体结构和生命周期的对象。 每个Yii应用系统只能包含一个应用主体,应用主体在 入口脚本中创建并能通过表达式 \Yii::$app 全局范围内访问。
补充: 当我们说"一个应用",它可能是一个应用主体对象,也可能是一个应用系统,是根据上下文来决定[译:中文为避免歧义,Application翻译为应
- assertThat用法
eksliang
JUnitassertThat
junit4.0 assertThat用法
一般匹配符1、assertThat( testedNumber, allOf( greaterThan(8), lessThan(16) ) );
注释: allOf匹配符表明如果接下来的所有条件必须都成立测试才通过,相当于“与”(&&)
2、assertThat( testedNumber, anyOf( g
- android点滴2
gundumw100
应用服务器android网络应用OSHTC
如何让Drawable绕着中心旋转?
Animation a = new RotateAnimation(0.0f, 360.0f,
Animation.RELATIVE_TO_SELF, 0.5f, Animation.RELATIVE_TO_SELF,0.5f);
a.setRepeatCount(-1);
a.setDuration(1000);
如何控制Andro
- 超简洁的CSS下拉菜单
ini
htmlWeb工作html5css
效果体验:http://hovertree.com/texiao/css/3.htmHTML文件:
<!DOCTYPE html>
<html xmlns="http://www.w3.org/1999/xhtml">
<head>
<title>简洁的HTML+CSS下拉菜单-HoverTree</title>
- kafka consumer防止数据丢失
kane_xie
kafkaoffset commit
kafka最初是被LinkedIn设计用来处理log的分布式消息系统,因此它的着眼点不在数据的安全性(log偶尔丢几条无所谓),换句话说kafka并不能完全保证数据不丢失。
尽管kafka官网声称能够保证at-least-once,但如果consumer进程数小于partition_num,这个结论不一定成立。
考虑这样一个case,partiton_num=2
- @Repository、@Service、@Controller 和 @Component
mhtbbx
DAOspringbeanprototype
@Repository、@Service、@Controller 和 @Component 将类标识为Bean
Spring 自 2.0 版本开始,陆续引入了一些注解用于简化 Spring 的开发。@Repository注解便属于最先引入的一批,它用于将数据访问层 (DAO 层 ) 的类标识为 Spring Bean。具体只需将该注解标注在 DAO类上即可。同时,为了让 Spring 能够扫描类
- java 多线程高并发读写控制 误区
qifeifei
java thread
先看一下下面的错误代码,对写加了synchronized控制,保证了写的安全,但是问题在哪里呢?
public class testTh7 {
private String data;
public String read(){
System.out.println(Thread.currentThread().getName() + "read data "
- mongodb replica set(副本集)设置步骤
tcrct
javamongodb
网上已经有一大堆的设置步骤的了,根据我遇到的问题,整理一下,如下:
首先先去下载一个mongodb最新版,目前最新版应该是2.6
cd /usr/local/bin
wget http://fastdl.mongodb.org/linux/mongodb-linux-x86_64-2.6.0.tgz
tar -zxvf mongodb-linux-x86_64-2.6.0.t
- rust学习笔记
wudixiaotie
学习笔记
1.rust里绑定变量是let,默认绑定了的变量是不可更改的,所以如果想让变量可变就要加上mut。
let x = 1; let mut y = 2;
2.match 相当于erlang中的case,但是case的每一项后都是分号,但是rust的match却是逗号。
3.match 的每一项最后都要加逗号,但是最后一项不加也不会报错,所有结尾加逗号的用法都是类似。
4.每个语句结尾都要加分

 3.文件准备
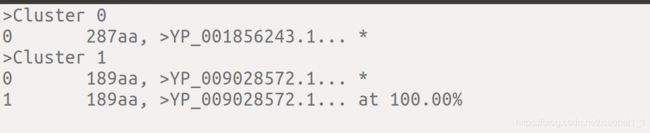
3.文件准备
 cd-hit运行时用很多参数可以进行调整设置,其运行命令为(参数仅为示例):
cd-hit运行时用很多参数可以进行调整设置,其运行命令为(参数仅为示例):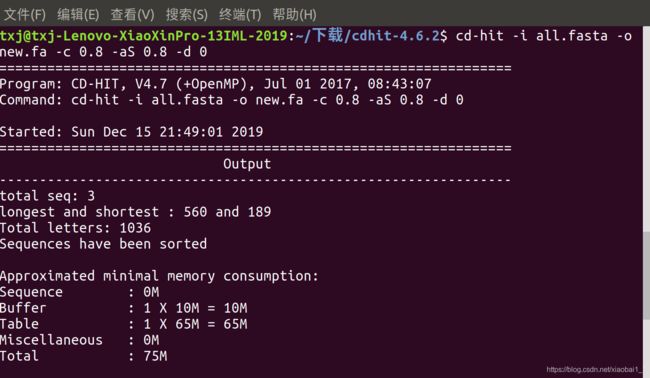
 格式首先以大于号“>”开头,接着是序列的标识符,然后是序列的描述信息。换行后是序列信息,标准核苷酸符号或氨基酸单字母符号。通常核苷酸符号大小写均可,而氨基酸一般用大写字母。文件中和每一行都不要超过80个字符(通常60个字符)。序列中允许空格,换行,空行,直到下一个大于号,表示该序列的结束。
格式首先以大于号“>”开头,接着是序列的标识符,然后是序列的描述信息。换行后是序列信息,标准核苷酸符号或氨基酸单字母符号。通常核苷酸符号大小写均可,而氨基酸一般用大写字母。文件中和每一行都不要超过80个字符(通常60个字符)。序列中允许空格,换行,空行,直到下一个大于号,表示该序列的结束。