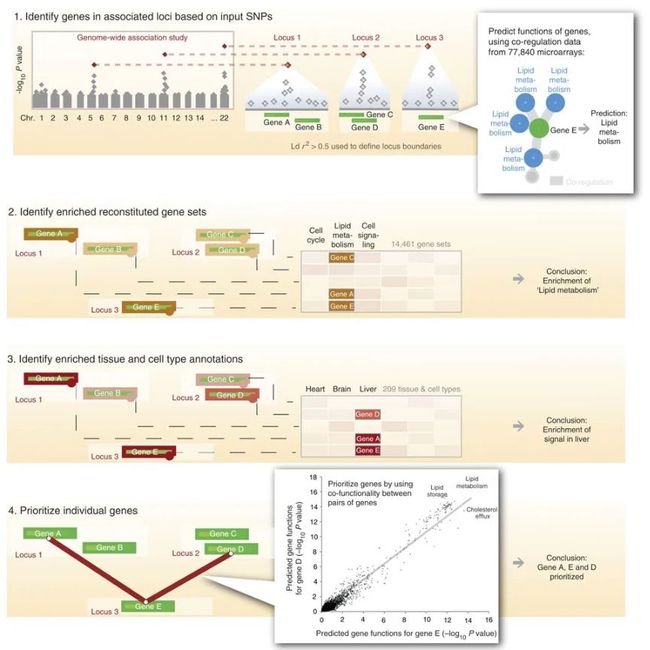
- 【DevOps】Backstage介绍及如何在Azure Kubernetes Service上进行部署
小涵
Azure云企业实践分享devopsazurekubernetes容器dockerbackstage
【DevOps】Backstage介绍及如何在AzureKubernetesService上进行部署推荐超级课程:本地离线DeepSeekAI方案部署实战教程【完全版】Docker快速入门到精通Kubernetes入门到大师通关课AWS云服务快速入门实战目录【DevOps】Backstage介绍及如何在AzureKubernetesService上进行部署Backstage介绍在AKS上部署Bac
- 二进制矩阵全零转换问题 | DFS
@Mr.stone
深度优先算法
问题描述在一个古老的实验室里,两个研究员,小星和小月,获得了一个mxn的电路图,表示为二进制矩阵grid。在这个矩阵中,他们可以对任意一个电路单元进行翻转操作。翻转操作会将所选单元的状态从0改为1,或从1改为0,同时影响与其相邻的上下左右单元。小星和小月希望通过最少的翻转次数,将整个电路图变成全0的状态。如果这个目标无法实现,则返回-1。测试样例样例1:输入:grid=[[0,1],[1,0]]输
- python 重构 Python 代码
隔壁小红馆
pythoncpythonpython面试pythoncpython
将for循环转换为list/dictionary/set表达式我们在时经常遇到的一个情况是,创建一个值的集合。比如我们创建一个列表,然后迭代地用值填充它,这里我们想创建一个立方数字的列表。大多数语言的标准方法如下:cubes=[]foriinrange(20):cubes.append(i**3)在Python中,我们可以使用列表表达式,生成需要的数据。就可以将代码简化为一行,省去定义列表,然后再
- python代码重构技巧_Python代码重构指南,老师Bryan Beecham完结
weixin_39916479
python代码重构技巧
本套课程由BryanBeecham,全球知名敏捷开发教练主讲的:Python代码重构指南。重构是软件改进的核心,它使软件拥有更好的结构和性能,也使代码更易于理解、修改和扩展。尽管重构并不是新事物,但是软件开发人员仍然会苦恼于如何正确地进行重构。随着敏捷运动的发展,DevOps之类的概念不断追求高质量和精心设计的代码,以实现更快的部署和反馈。不过,现有的很多关于重构的教程都基于Java语言,关于Py
- python语言对代码的块结构不敏感_浅谈python(二)--python代码规范
初夏之菡
对于每一门语言来说,都有自己的编码规则,编程时是不可以违背这些准则的,一旦不遵守这个准则,程序就会报错无法执行,本节将介绍下python的一些编码规则。1、代码缩进与冒号首先介绍下代码缩进有什么用处,代码缩进是指通过在一行代码的前输入若干空格或者制表符来表示行与行之间的层次关系,每一种编程语言一般都需要代码缩进进行规范程序代码的层次结构,让代码清晰易于解读。对于其它的语言来说,代码缩进作为一种良好
- Python编码系列—Python代码重构:提升代码质量
学步_技术
Python编码python重构开发语言
欢迎来到我的技术小筑,一个专为技术探索者打造的交流空间。在这里,我们不仅分享代码的智慧,还探讨技术的深度与广度。无论您是资深开发者还是技术新手,这里都有一片属于您的天空。让我们在知识的海洋中一起航行,共同成长,探索技术的无限可能。探索专栏:学步_技术的首页——持续学习,不断进步,让学习成为我们共同的习惯,让总结成为我们前进的动力。技术导航:人工智能:深入探讨人工智能领域核心技术。自动驾驶:分享自动
- 【python】函数重构
划过手的泪滴t
python每日一练云计算运维python重构服务器开发语言每日一练运维
函数重构函数重构pycharm函数重构步骤函数重构练习函数重构函数重构是指对现有函数进行修改和优化的过程。重构的目的是改善代码的可读性、可维护性和灵活性,同时保持其功能不变。函数重构通常包括以下步骤:理解函数的功能和目的。了解函数的作用和期望结果,确定重构的目标。检查函数的代码质量。查看函数的代码是否清晰、简洁、可读,有无可改进之处。提取重复的代码。如果函数中有重复的代码块,可以将其提取为单独的函
- K8S之QoS详解
RedCong
云原生k8sOpenshiftkubernetes容器云原生
PodQoS类服务质量(QualityofService,QoS)类,阐述Kubernetes如何根据为Pod中的容器指定的资源约束为每个Pod设置QoS类。Kubernetes依赖这种分类来决定当Node上没有足够可用资源时要驱逐哪些Pod。QoS类(QualityofServiceclasses)Kubernetes对你运行的Pod进行分类,并将每个Pod分配到特定的QoS类中。Kuberne
- Python入门实战:Python的代码重构
AI智能涌现深度研究
DeepSeekR1&大数据AI人工智能大数据人工智能语言模型AILLMJavaPython架构设计
1.背景介绍Python是一种基于社区发展、易用性、生态系统完善、可扩展性强、性能卓越等特点的高级编程语言。作为一门解释型语言,它具有高效率、简洁语法、丰富的库函数、跨平台能力和多种开发范式等优点。但随着项目不断迭代更新,代码量逐渐增加,导致代码结构混乱、缺乏模块化设计、重复逻辑过多、命名不规范等问题。如何有效地组织、管理和维护代码、提升代码质量、更好地实现功能,是一个技术人的日常工作。如何进行代
- python 使用microsoft-Florence-2-base进行图片描述生成
哦里 哦里哦里给
AI大语言模型实战pythonmicrosoft开发语言
目录一、Florence-2简介二、代码实践三、多语言模型一、Florence-2简介Florence-2是一个先进的视觉基础模型,采用基于提示(prompt)的方式,处理广泛的视觉和视觉-语言任务。Florence-2能够解析简单的文本提示,执行如图像描述、物体检测和分割等任务。该模型利用FLD-5B数据集,该数据集包含54亿个注释,涵盖1.26亿张图像,用于掌握多任务学习。模型的序列到序列架构
- MDX语言的设备管理
穆骊瑶
包罗万象golang开发语言后端
设备管理中的MDX语言应用引言设备管理是在各行各业中都至关重要的一环,尤其是在制造业、物流业、以及信息技术等领域。设备的正常运行直接关系到企业的生产效率和经济效益。随着信息技术的不断发展,现代企业越来越依赖数据来优化设备管理。而MDX(MultidimensionalExpressions)语言作为多维数据库查询的标准语言,能够有效支持设备管理中的数据分析和决策支持。本文将深入探讨MDX语言在设备
- 从0到1构建AI深度学习视频分析系统--基于YOLO 目标检测的动作序列检查系统:(2)消息队列与消息中间件
shiter
人工智能系统解决方案与技术架构人工智能深度学习音视频
文章大纲原始视频队列Python内存视频缓存优化方案(4GB以内)一、核心参数设计二、内存管理实现三、性能优化策略四、内存占用验证五、高级优化技巧六、部署建议检测结果队列YOLO检测结果队列技术方案一、技术选型矩阵二、核心实现代码三、性能优化策略四、可视化方案对比五、部署建议逻辑判定队列时间片图论时间序列大模型引入参考文献原始视频队列想要在单机内存中缓存1-5分钟的视频片段,python技术栈的话
- 【esp32】VSCODE + esp-idf 使用记录
zscredstone
vscodeide编辑器
旨在进行学习使用过程中的问题记录。esp已经把vscode插件做的不错了,可以直接进行编译调试。使用的是esp32S3内置的usb/jtag主要参考:https://blog.csdn.net/weixin_50993868/article/details/136498570https://blog.csdn.net/weixin_43842462/article/details/12329584
- Python--操作系统进行交互 【OS库】
~请叫我小祸害~
python开发语言
在Python中,os是一个内置的标准库,用于与操作系统进行交互。它提供了许多函数和方法,用于执行与操作系统相关的任务,例如文件类操作和目录操作、进程管理、环境变量访问等。接下来我给大家列举一下比较常用的文件操作方法:代码示例⬇⬇⬇⬇⬇⬇⬇:1、os.getcwd():返回当前工作目录的路径。importos#返回当前工作目录current_dir=os.getcwd()print("当前工作目录
- C#请求队列后台服务
~请叫我小祸害~
.NET/C#c#jvm开发语言
队列执行1、首先需要一个公共类里面有我们的队列需要执行的方法这个方法最好是一个单独的不受别的控制器影响的class。因为你如果声明错位置的话一不小心就会把我们的队列重置。导致队列内容丢失造成损失。大家如果有某些方法运行时间太长但又需要快速给出结果,就可以利用队列,我们这边直接给她结果。然后具体操作我们在内部慢慢执行。//比如说我这里新建一个Class专门用来存放这个方法和队列内容容器publicc
- 10 个极其有用的 Python 自动化脚本
python
在现代职场中,重复性和耗时的任务常常占据大量时间,影响工作效率。Python作为一种高效、易用的编程语言,提供了丰富的库和工具,能够帮助打工人自动化处理日常任务,提升工作效率。以下是十个必备的Python自动化脚本:一、文件批量重命名脚本在日常工作中,可能需要对大量文件进行重命名操作。手动操作既耗时又容易出错。使用Python脚本,可以实现文件的批量重命名,提高效率。importosdefbatc
- python内存泄露
weixin_39810989
内存泄漏pythonmalloc
定位工具及使用1.tracemalloc可以通过创建快照的方式记录当前的内存占用情况。从而可以比较快照与快照之间的内存占用差异。可以获取内存块的回溯,定位到内存占用最多的文件和代码行。osgeo.cn/cpython/library/tracemalloc.html2.pympler可以创建快照(summaries)进行内存块占用对比frompympler.classtrackerimportCl
- FreeModbus V1.6 主机使用说明(二)
七七知享
Modbusstm32嵌入式硬件单片机mcu物联网开发语言算法
二、移植对于协议栈的移植主要包括两个方面,硬件及软件。用户需要根据自己的需求进行自行选择。注:以下所有说明都主要针对Modbus主机模式进行介绍。2.1、软件软件方面支持基于裸机及实时操作系统的移植;支持单个主机与单个从机同时独立运行。另外用户也可以修改协议栈的事件回调接口,使主机请求的接口采用阻塞及非阻塞模式;主机资源等待方面,用户也可以设置等待超时时间等等,诸多功能将会一一介绍。2.1.1、操
- 金融时间序列分析(Yahoo Finance API实战)
闲人编程
Python数据分析实战精要金融yfinance时间序列波动率数据归一化数据分析Dash
这里写目录标题金融时间序列分析(YahooFinanceAPI实战)1.引言2.项目背景与意义3.数据集介绍4.GPU加速在数据处理中的应用5.交互式GUI设计与加速处理6.系统整体架构7.数学公式与指标计算8.完整代码实现9.代码自查与BUG排查10.总结与展望金融时间序列分析(YahooFinanceAPI实战)1.引言在当今金融市场中,时间序列数据分析是理解股票、指数以及其他金融产品走势的重
- 系分 02 软件工程
一越王超
软考系统分析师软件工程
软件工程本身涵盖内容很广,从系统规划到分析……到维护都属于软件工程,但是我们将会在其他章节讨论相关内容,本节我们主要内容如下:系统规划软件工程信息系统生命周期(★)软件开发模型(★★★★)逆向工程(★★)净室软件工程(★)需求工程系统设计系统测试与维护基础知识软件工程是指应用计算机科学、数学及管理科学等原理,以工程化的原则和方法来解决软件问题的工程,其目的是提高软件生产率、提高软件质量、减低软件成
- 区块链架构、跨链和演进
Omni-Space
区块链(BlockChain)区块链架构跨链演进
本文是基于作者近几年来对各种区块链平台理念和技术的研究,结合作者过去十多年的IT经验,审慎思考的结果,文章仅代表作者个人观点。作者会假设读者对各种区块链平台有一定的认知,不会对具体的区块链平台再做详细的介绍。为了从根本上说清楚区块链的架构内涵,作者先概括出区块链的本质,从区块链的本质出发,以发展的眼光给出一个区块链的详见附件架构,并对高阶的各个模块进行详细的说明。还会从区块链跨链的本质出发,说明区
- 洛谷-P5534 【XR-3】等差数列
兔子递归
洛谷题解c++经验分享
题目:P5534【XR-3】等差数列题目分析:首先得输入,然后根据前两项的值算出“d”,即a[2]-a[1]。提示:十年OI一场空,不开longlong见祖宗接着求出前n项:从3开始,到n项结束。递推公式:"a[i]=a[i-1]+d;"。最后从1到n累加,输出。上代码:#includeusingnamespacestd;longa[1000005];intmain(void){longi,d,a
- 【从零开始学习计算机科学】软件工程(三)需求工程
贫苦游商
学习软件工程需求分析软件需求需求文档软件开发敏捷编程
【从零开始学习计算机科学】软件工程(三)需求工程需求工程好的需求应具备的特征:需求工程(RequirementEngineering,RE)起始导出需求讨论会头脑风暴调查问卷场景分析法实地考察原型法精化协商规格说明确认需求管理需求工程设计和开发一个计算机软件时,如果软件解决的问题不对,那么再精巧的软件也满足不了任何人的要求。理解问题的需求是软件工程师所面对的最困难的任务之一。困难的原因有二:客户不
- 【从零开始学习计算机科学】软件工程(五)软件设计
贫苦游商
学习软件工程软件开发软件设计敏捷开发极限编程软件需求
【从零开始学习计算机科学】软件工程(五)软件设计软件设计概述良好的设计具有三大特性设计主要包含的方面设计中的一些概念设计的方法与策略体系结构设计体系结构设计的基本问题:体系结构的设计模式体系结构设计的过程构建级设计面向对象构件设计用户接口设计用户接口设计原则:用户接口分析的目标:设计的评审软件设计概述软件的分析偏重于问题域,描述软件要做什么,而设计则偏重于解决方案,描述软件究竟要如何做。设计创建了
- 【从零开始学习计算机科学】软件工程(二)软件工程方法学
贫苦游商
学习软件工程hadoop面向过程面向对象软件开发敏捷开发
【从零开始学习计算机科学】软件工程(二)软件工程方法学软件工程方法学结构化/面向过程结构化编程结构化设计结构化分析结构化方法的常见问题面向对象软件工程方法学我们通常把在软件生命周期全过程中使用的一整套技术方法的集合称为方法学(methodology),也称为范型(paradigm)。软件工程中有许多方法:结构化/面向过程对于结构化方法,其又被称为传统方法学,也称为生命周期方法学或结构化范型。它采用
- 【从零开始学习计算机科学】数字逻辑(四)数字系统设计
贫苦游商
学习数字逻辑verilog数字系统HDL数字电路FPGA
【从零开始学习计算机科学】数字逻辑(四)数字系统设计数字系统设计硬件描述语言HDL(HardwareDescriptionLanguage)VerilogHDL的起源与发展HDL软核、固核和硬核的重用HDL的应用数字系统设计实现数字系统设计一个数字集成电路的可以从不同的层次(系统级、算法级、寄存器传输级、门级、开关级)以及不同的领域(行为领域、结构领域、物理领域)进行描述。三个领域主要含义如下:行
- c#中将数据库数据导出到EXCEL中
lujunql
技术excel数据库c#microsoftlibrarystring
我分以下几步进行介绍:1,新建一个C#应用程序,在对话框上放置一个按钮,Name=buttonOutput,Text=Output,用这个按钮激发导出程序;2,添加对“MicrosoftExcel9.0ObjectLibrary”的引用,根据自己计算机上安装Office版本的来确定Library的版本;3,在代码中加入引用:usingExcel;usingSystem.Reflection;4,在
- Selenium时间等待_显示等待
Mr_Python2024
seleniumpython
特点:针对具体元素进行时间等待可以自定义等待时长和间隔时间按照设定的时间,不断定位元素,定位到了直接执行下一步操作如在设定时间内没定位到元素,则报错(TimeOutException)显示等待概念:设置一个等待时间和一个条件,在规定时间内,每隔一段时间查看下条件是否成立,如果成立那么程序就继续执行,否则就提示一个超时异常(TimeoutException)。在使用显示等待时;需要结合Seleniu
- 在Python中如何检测和解决内存泄漏问题
python资深爱好者
pythonjvm
在Python中,内存泄漏通常不是像在一些低级语言(如C或C++)中那样常见,因为Python的内存管理(包括自动垃圾回收)相对高级且自动化。然而,在长时间运行的应用程序中,特别是在使用大量循环、大型数据结构或外部库时,仍然可能出现内存泄漏。以下是在Python中检测和解决内存泄漏的一些方法:1.使用内存分析工具a.objgraphobjgraph是一个用于分析Python对象图的库,可以帮助你识
- Python在数据处理中的应用:从入门到精通
程之编
python信息可视化开发语言
活动发起人@小虚竹想对你说:这是一个以写作博客为目的的创作活动,旨在鼓励大学生博主们挖掘自己的创作潜能,展现自己的写作才华。如果你是一位热爱写作的、想要展现自己创作才华的小伙伴,那么,快来参加吧!我们一起发掘写作的魅力,书写出属于我们的故事。我们诚挚邀请你参加为期14天的创作挑战赛!在当今数字化时代,数据处理已成为各个领域不可或缺的一部分。无论是企业决策、科学研究还是日常的个人数据分析,掌握高效的
- jsonp 常用util方法
hw1287789687
jsonpjsonp常用方法jsonp callback
jsonp 常用java方法
(1)以jsonp的形式返回:函数名(json字符串)
/***
* 用于jsonp调用
* @param map : 用于构造json数据
* @param callback : 回调的javascript方法名
* @param filters : <code>SimpleBeanPropertyFilter theFilt
- 多线程场景
alafqq
多线程
0
能不能简单描述一下你在java web开发中需要用到多线程编程的场景?0
对多线程有些了解,但是不太清楚具体的应用场景,能简单说一下你遇到的多线程编程的场景吗?
Java多线程
2012年11月23日 15:41 Young9007 Young9007
4
0 0 4
Comment添加评论关注(2)
3个答案 按时间排序 按投票排序
0
0
最典型的如:
1、
- Maven学习——修改Maven的本地仓库路径
Kai_Ge
maven
安装Maven后我们会在用户目录下发现.m2 文件夹。默认情况下,该文件夹下放置了Maven本地仓库.m2/repository。所有的Maven构件(artifact)都被存储到该仓库中,以方便重用。但是windows用户的操作系统都安装在C盘,把Maven仓库放到C盘是很危险的,为此我们需要修改Maven的本地仓库路径。
- placeholder的浏览器兼容
120153216
placeholder
【前言】
自从html5引入placeholder后,问题就来了,
不支持html5的浏览器也先有这样的效果,
各种兼容,之前考虑,今天测试人员逮住不放,
想了个解决办法,看样子还行,记录一下。
【原理】
不使用placeholder,而是模拟placeholder的效果,
大概就是用focus和focusout效果。
【代码】
<scrip
- debian_用iso文件创建本地apt源
2002wmj
Debian
1.将N个debian-506-amd64-DVD-N.iso存放于本地或其他媒介内,本例是放在本机/iso/目录下
2.创建N个挂载点目录
如下:
debian:~#mkdir –r /media/dvd1
debian:~#mkdir –r /media/dvd2
debian:~#mkdir –r /media/dvd3
….
debian:~#mkdir –r /media
- SQLSERVER耗时最长的SQL
357029540
SQL Server
对于DBA来说,经常要知道存储过程的某些信息:
1. 执行了多少次
2. 执行的执行计划如何
3. 执行的平均读写如何
4. 执行平均需要多少时间
列名 &
- com/genuitec/eclipse/j2eedt/core/J2EEProjectUtil
7454103
eclipse
今天eclipse突然报了com/genuitec/eclipse/j2eedt/core/J2EEProjectUtil 错误,并且工程文件打不开了,在网上找了一下资料,然后按照方法操作了一遍,好了,解决方法如下:
错误提示信息:
An error has occurred.See error log for more details.
Reason:
com/genuitec/
- 用正则删除文本中的html标签
adminjun
javahtml正则表达式去掉html标签
使用文本编辑器录入文章存入数据中的文本是HTML标签格式,由于业务需要对HTML标签进行去除只保留纯净的文本内容,于是乎Java实现自动过滤。
如下:
public static String Html2Text(String inputString) {
String htmlStr = inputString; // 含html标签的字符串
String textSt
- 嵌入式系统设计中常用总线和接口
aijuans
linux 基础
嵌入式系统设计中常用总线和接口
任何一个微处理器都要与一定数量的部件和外围设备连接,但如果将各部件和每一种外围设备都分别用一组线路与CPU直接连接,那么连线
- Java函数调用方式——按值传递
ayaoxinchao
java按值传递对象基础数据类型
Java使用按值传递的函数调用方式,这往往使我感到迷惑。因为在基础数据类型和对象的传递上,我就会纠结于到底是按值传递,还是按引用传递。其实经过学习,Java在任何地方,都一直发挥着按值传递的本色。
首先,让我们看一看基础数据类型是如何按值传递的。
public static void main(String[] args) {
int a = 2;
- ios音量线性下降
bewithme
ios音量
直接上代码吧
//second 几秒内下降为0
- (void)reduceVolume:(int)second {
KGVoicePlayer *player = [KGVoicePlayer defaultPlayer];
if (!_flag) {
_tempVolume = player.volume;
- 与其怨它不如爱它
bijian1013
选择理想职业规划
抱怨工作是年轻人的常态,但爱工作才是积极的心态,与其怨它不如爱它。
一般来说,在公司干了一两年后,不少年轻人容易产生怨言,除了具体的埋怨公司“扭门”,埋怨上司无能以外,也有许多人是因为根本不爱自已的那份工作,工作完全成了谋生的手段,跟自已的性格、专业、爱好都相差甚远。
- 一边时间不够用一边浪费时间
bingyingao
工作时间浪费
一方面感觉时间严重不够用,另一方面又在不停的浪费时间。
每一个周末,晚上熬夜看电影到凌晨一点,早上起不来一直睡到10点钟,10点钟起床,吃饭后玩手机到下午一点。
精神还是很差,下午像一直野鬼在城市里晃荡。
为何不尝试晚上10点钟就睡,早上7点就起,时间完全是一样的,把看电影的时间换到早上,精神好,气色好,一天好状态。
控制让自己周末早睡早起,你就成功了一半。
有多少个工作
- 【Scala八】Scala核心二:隐式转换
bit1129
scala
Implicits work like this: if you call a method on a Scala object, and the Scala compiler does not see a definition for that method in the class definition for that object, the compiler will try to con
- sudoku slover in Haskell (2)
bookjovi
haskellsudoku
继续精简haskell版的sudoku程序,稍微改了一下,这次用了8行,同时性能也提高了很多,对每个空格的所有解不是通过尝试算出来的,而是直接得出。
board = [0,3,4,1,7,0,5,0,0,
0,6,0,0,0,8,3,0,1,
7,0,0,3,0,0,0,0,6,
5,0,0,6,4,0,8,0,7,
- Java-Collections Framework学习与总结-HashSet和LinkedHashSet
BrokenDreams
linkedhashset
本篇总结一下两个常用的集合类HashSet和LinkedHashSet。
它们都实现了相同接口java.util.Set。Set表示一种元素无序且不可重复的集合;之前总结过的java.util.List表示一种元素可重复且有序
- 读《研磨设计模式》-代码笔记-备忘录模式-Memento
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.util.ArrayList;
import java.util.List;
/*
* 备忘录模式的功能是,在不破坏封装性的前提下,捕获一个对象的内部状态,并在对象之外保存这个状态,为以后的状态恢复作“备忘”
- 《RAW格式照片处理专业技法》笔记
cherishLC
PS
注意,这不是教程!仅记录楼主之前不太了解的
一、色彩(空间)管理
作者建议采用ProRGB(色域最广),但camera raw中设为ProRGB,而PS中则在ProRGB的基础上,将gamma值设为了1.8(更符合人眼)
注意:bridge、camera raw怎么设置显示、输出的颜色都是正确的(会读取文件内的颜色配置文件),但用PS输出jpg文件时,必须先用Edit->conv
- 使用 Git 下载 Spring 源码 编译 for Eclipse
crabdave
eclipse
使用 Git 下载 Spring 源码 编译 for Eclipse
1、安装gradle,下载 http://www.gradle.org/downloads
配置环境变量GRADLE_HOME,配置PATH %GRADLE_HOME%/bin,cmd,gradle -v
2、spring4 用jdk8 下载 https://jdk8.java.
- mysql连接拒绝问题
daizj
mysql登录权限
mysql中在其它机器连接mysql服务器时报错问题汇总
一、[running]
[email protected]:~$mysql -uroot -h 192.168.9.108 -p //带-p参数,在下一步进行密码输入
Enter password: //无字符串输入
ERROR 1045 (28000): Access
- Google Chrome 为何打压 H.264
dsjt
applehtml5chromeGoogle
Google 今天在 Chromium 官方博客宣布由于 H.264 编解码器并非开放标准,Chrome 将在几个月后正式停止对 H.264 视频解码的支持,全面采用开放的 WebM 和 Theora 格式。
Google 在博客上表示,自从 WebM 视频编解码器推出以后,在性能、厂商支持以及独立性方面已经取得了很大的进步,为了与 Chromium 现有支持的編解码器保持一致,Chrome
- yii 获取控制器名 和方法名
dcj3sjt126com
yiiframework
1. 获取控制器名
在控制器中获取控制器名: $name = $this->getId();
在视图中获取控制器名: $name = Yii::app()->controller->id;
2. 获取动作名
在控制器beforeAction()回调函数中获取动作名: $name =
- Android知识总结(二)
come_for_dream
android
明天要考试了,速速总结如下
1、Activity的启动模式
standard:每次调用Activity的时候都创建一个(可以有多个相同的实例,也允许多个相同Activity叠加。)
singleTop:可以有多个实例,但是不允许多个相同Activity叠加。即,如果Ac
- 高洛峰收徒第二期:寻找未来的“技术大牛” ——折腾一年,奖励20万元
gcq511120594
工作项目管理
高洛峰,兄弟连IT教育合伙人、猿代码创始人、PHP培训第一人、《细说PHP》作者、软件开发工程师、《IT峰播》主创人、PHP讲师的鼻祖!
首期现在的进程刚刚过半,徒弟们真的很棒,人品都没的说,团结互助,学习刻苦,工作认真积极,灵活上进。我几乎会把他们全部留下来,现在已有一多半安排了实际的工作,并取得了很好的成绩。等他们出徒之日,凭他们的能力一定能够拿到高薪,而且我还承诺过一个徒弟,当他拿到大学毕
- linux expect
heipark
expect
1. 创建、编辑文件go.sh
#!/usr/bin/expect
spawn sudo su admin
expect "*password*" { send "13456\r\n" }
interact
2. 设置权限
chmod u+x go.sh 3.
- Spring4.1新特性——静态资源处理增强
jinnianshilongnian
spring 4.1
目录
Spring4.1新特性——综述
Spring4.1新特性——Spring核心部分及其他
Spring4.1新特性——Spring缓存框架增强
Spring4.1新特性——异步调用和事件机制的异常处理
Spring4.1新特性——数据库集成测试脚本初始化
Spring4.1新特性——Spring MVC增强
Spring4.1新特性——页面自动化测试框架Spring MVC T
- idea ubuntuxia 乱码
liyonghui160com
1.首先需要在windows字体目录下或者其它地方找到simsun.ttf 这个 字体文件。
2.在ubuntu 下可以执行下面操作安装该字体:
sudo mkdir /usr/share/fonts/truetype/simsun
sudo cp simsun.ttf /usr/share/fonts/truetype/simsun
fc-cache -f -v
- 改良程序的11技巧
pda158
技巧
有很多理由都能说明为什么我们应该写出清晰、可读性好的程序。最重要的一点,程序你只写一次,但以后会无数次的阅读。当你第二天回头来看你的代码 时,你就要开始阅读它了。当你把代码拿给其他人看时,他必须阅读你的代码。因此,在编写时多花一点时间,你会在阅读它时节省大量的时间。
让我们看一些基本的编程技巧:
尽量保持方法简短
永远永远不要把同一个变量用于多个不同的
- 300个涵盖IT各方面的免费资源(下)——工作与学习篇
shoothao
创业免费资源学习课程远程工作
工作与生产效率:
A. 背景声音
Noisli:背景噪音与颜色生成器。
Noizio:环境声均衡器。
Defonic:世界上任何的声响都可混合成美丽的旋律。
Designers.mx:设计者为设计者所准备的播放列表。
Coffitivity:这里的声音就像咖啡馆里放的一样。
B. 避免注意力分散
Self Co
- 深入浅出RPC
uule
rpc
深入浅出RPC-浅出篇
深入浅出RPC-深入篇
RPC
Remote Procedure Call Protocol
远程过程调用协议
它是一种通过网络从远程计算机程序上请求服务,而不需要了解底层网络技术的协议。RPC协议假定某些传输协议的存在,如TCP或UDP,为通信程序之间携带信息数据。在OSI网络通信模型中,RPC跨越了传输层和应用层。RPC使得开发