分子对接结果分析和作图
原文链接:https://mp.weixin.qq.com/s/D5eLXvCUAln2NOj2fDw3vg
分子对接是最常用的分子模拟工具,用来探究任何有相互作用的生物活性分子之间的相互作用细节。然而对接产生的多个可能的构象到底那个是天然存在的构象?确定了构象,什么作用力,作用位点起到关键作用?
确定最佳构象,有两种根据,一是binding pose最多的,一种是打分函数评价最好的。
然而这两个如果是一类结合构象(如RMSD<2.0A),那么可以50%(属个人经验,没有根据)以上确定这个结合构象为最佳的。如果出现模棱两可,根据已有的pdb数据库数据或者参考文献确定(对自己蛋白结构的了解程度)。
归纳起来即:
(1)First filter/ranking is usually the score of the pose. As the scoring function is a problem by itself for docking protocol,.
(2)RMSD and the number of interacting residues and different type of interactions(hydrogen bonding,Hydrophobic,Ionic,Aromatic,Cation-π)
(3)Third is visual or empirical analysis analyzing : if you have a structure of your protein with a ligand, try to check if it is consistent. If not, check that the interactions you see are good.
那么怎样看相互作用的细节或者作用力呢?
推荐3个2D工具,以蛋白配体为例,2D图表明配体与蛋白的那些氨基酸有明确的相互作用,作用方式是什么等信息。如图所示,分别是由Ligplot,poseview和DS 4.5绘制的蛋白与辅酶NAD+的相互作用2D图:
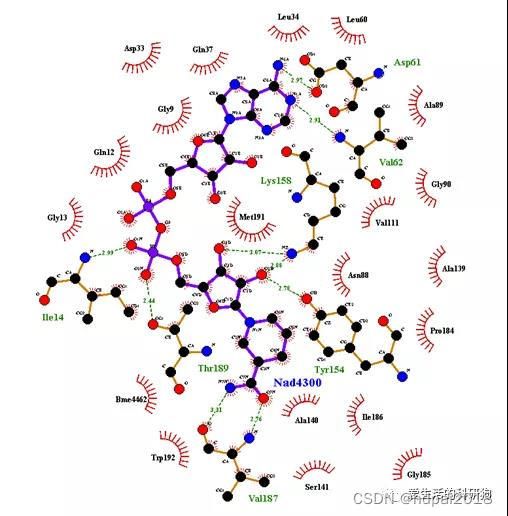
Interaction between NAD+ and the enzyme binding site. H-bonds are displayed as green dashed lines. Hydrophobic(or non-bonded ) interactions are indicated as red opposite arcs.
Poseview:PDB数据库中分析蛋白和共结晶配体相互作用就用的是poseview,个人感觉是最漂亮的2D图,然而并不免费,这儿有一个网站,http://poseview.zbh.uni-hamburg.de/
可以做出poseview图,但是只能分析配体蛋白之间的氢键作用。

Interaction between NAD+ and the enzyme binding site. The interaction pattern is composed of hydrogen bonds, visualized as black dashed lines; π interactions, shown as green dashed lines with dots denoting the participating π systems; and hydrophobic contacts, which are represented by the residue labels and spline segments along the contacting hydrophobic ligand parts.

The cofactor-binding site. The key interactions, The related polar interactions and H-bond patterns between NAD+ and enzyme.
2个3D工具,Chimera 和pymol。
Chimera比较好的一点是把结果拖进去就可以展示出相互作用的氨基酸残基,非常方便。
Pymol 可展示出配体蛋白的氢键作用,find ------ polar contacts。
(1)这些软件在linux or windows 7/8/10安装使用;
(2)如何对接及对接结果分析;
(3)更好用的工具