monocle2轨迹分析
<生信交流与合作请关注公众号@生信探索>
安装monocle2
- 下载源代码
wget https://www.bioconductor.org/packages/release/bioc/src/contrib/monocle_2.26.0.tar.gz
tar -xf monocle*gz
- 修改源代码bug
monocle/R/BEAM.R
#203行的
progenitor_method == 'duplicate'
#改为
'duplicate' %in% progenitor_method
#251行的
progenitor_method == 'sequential_split'
#改为
'sequential_split' %in% progenitor_method
monocle/R/order_cells.R
#删除1620行
if(class(projection) != 'matrix')
- 安装
install.packages(remotes)
library(remotes)
remotes::install_local("monocle")
从anndata导出monocle所需的输入文件
bdata是注释好细胞类型的anndata对象
adata是原始的count数据,没经过任何处理
所以要从bdata获得pd,adata.var作为fd,adata.X作为count信息
import pandas as pd
import scanpy as sc
from scipy.io import mmwrite
adata=sc.datasets.pbmc3k()
bdata=sc.datasets.pbmc3k_processed()
adata
# AnnData object with n_obs × n_vars = 2700 × 32738
# var: 'gene_ids'
bdata
# AnnData object with n_obs × n_vars = 2638 × 1838
# obs: 'n_genes', 'percent_mito', 'n_counts', 'louvain'
# var: 'n_cells'
# uns: 'draw_graph', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
# obsm: 'X_pca', 'X_tsne', 'X_umap', 'X_draw_graph_fr'
# varm: 'PCs'
# obsp: 'distances', 'connectivities'
- pd
adata = adata[adata.obs_names.isin(bdata.obs_names)]
m=pd.merge(adata.obs,bdata.obs,left_index=True,right_index=True,how="left")
adata.obs = m.loc[:,["louvain"]]
- fd 删除不表达的基因
lg=adata.X.sum(axis=0)>0.5
lg=lg.tolist()[0]
adata=adata[:,lg]
- 修改列名
adata.obs.rename(columns={"louvain":"cell_type"},inplace=True)
adata.obs.insert(loc=0,column="Obs",value=adata.obs_names)
adata.var.insert(loc=0,column="gene_short_name",value=adata.var_names)
- 保存数据
mmwrite("PBMC.mtx",adata.X.transpose())
adata.obs.to_csv("PBMC_pd.csv",index=False)
adata.var.loc[:,["gene_short_name"]].to_csv("PBMC_fd.csv",index=False)
- 高变基因
sc.pp.normalize_total(adata, target_sum=10000, inplace=True)
sc.pp.log1p(adata,base=None)
sc.pp.highly_variable_genes(adata,n_top_genes=2000,flavor="seurat", subset=True,inplace=True)
pd.DataFrame({"Var":adata.var_names}).to_csv("PBMC_hvg.csv",index=False)
- 得到如下4个文件
PBMC.mtx
PBMC_fd.csv
PBMC_hvg.csv
PBMC_pd.csv
- 压缩mtxa文件
gzip PBMC.mtx
- 文件内容
# PBMC_fd.csv
gene_short_name
AL627309.1
AP006222.2
#PBMC_hvg.csv
Var
TNFRSF4
CPSF3L
#PBMC_pd.csv
Obs,cell_type
AAACATACAACCAC-1,CD4 T cells
AAACATTGAGCTAC-1,B cells
构造monocle对象
suppressPackageStartupMessages(library(data.table))
suppressPackageStartupMessages(library(tibble))
suppressPackageStartupMessages(library(dplyr))
suppressPackageStartupMessages(library(ggplot2))
suppressPackageStartupMessages(library(tidyr))
suppressPackageStartupMessages(library(magrittr))
suppressPackageStartupMessages(library(patchwork))
suppressPackageStartupMessages(library(Matrix))
suppressPackageStartupMessages(library(monocle))
data <- Matrix::readMM("PBMC.mtx.gz")
pd <- data.table::fread("PBMC_pd.csv", data.table = FALSE, header = TRUE) %>%
tibble::column_to_rownames("Obs")
fd <- data.table::fread("PBMC_fd.csv", data.table = FALSE, header = TRUE)
rownames(fd) <- fd$gene_short_name
cds <- newCellDataSet(data,
phenoData = new("AnnotatedDataFrame", data = pd),
featureData = new("AnnotatedDataFrame", data = fd),
expressionFamily = negbinomial.size()
)
rm(data,pd,fd) # 删除不需要的变量
估计size factor和离散度
cds <- estimateSizeFactors(cds)
cds <- estimateDispersions(cds)
轨迹定义基因选择及可视化和构建轨迹
ordering_genes <- data.table::fread("PBMC_hvg.csv", header = TRUE) %>%
dplyr::pull(1)
ordering_genes <- intersect(rownames(cds), ordering_genes)
cds <- setOrderingFilter(cds, ordering_genes)
plot_ordering_genes(cds)
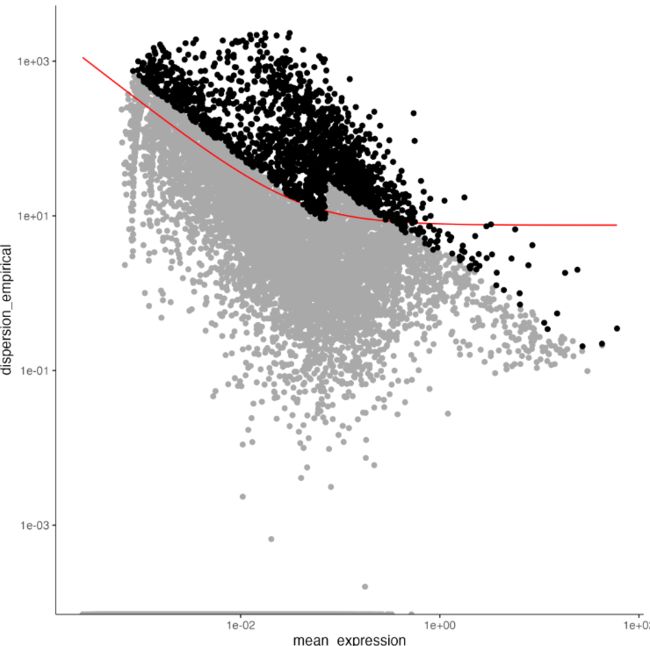
降维
cds <- reduceDimension(cds, max_components = 2, method = "DDRTree")
cds <- orderCells(cds)
轨迹图
#以pseudotime值上色
plot_cell_trajectory(cds,color_by="Pseudotime", size=1,show_backbone=TRUE)
#以细胞类型上色
plot_cell_trajectory(cds,color_by="cell_type", size=1,show_backbone=TRUE)
#以细胞状态上色
plot_cell_trajectory(cds, color_by = "State",size=1,show_backbone=TRUE)
#轨迹图分面显示
plot_cell_trajectory(cds, color_by = "State") + facet_wrap("~State", nrow = 1)
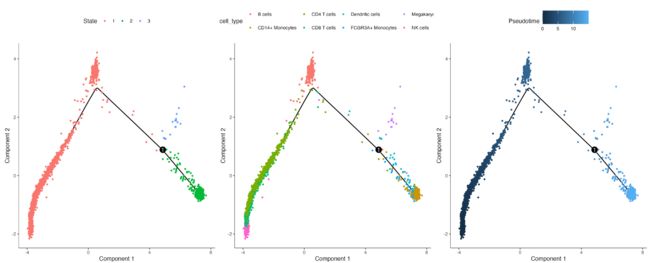
树状图
p1 <- plot_cell_trajectory(mycds, x = 1, y = 2, color_by = "cell_type") +
theme(legend.position='none',panel.border = element_blank()) #去掉第一个的legend
p2 <- plot_complex_cell_trajectory(mycds, x = 1, y = 2,
color_by = "cell_type")+
theme(legend.title = element_blank())
p1|p2
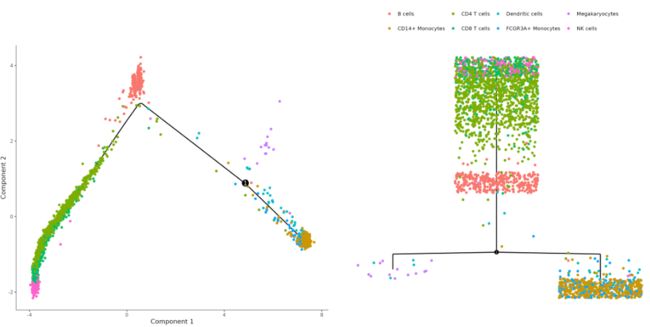
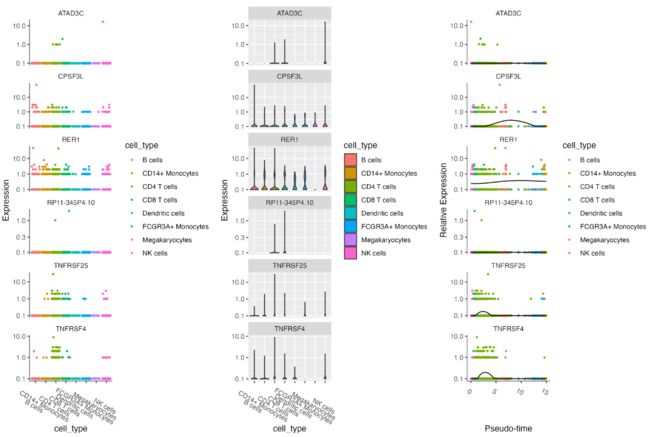
指定基因的可视化
s.genes <- c("需要可视化的基因向量")
p1 <- plot_genes_jitter(cds[s.genes,], grouping = "cell_type", color_by = "cell_type")
p2 <- plot_genes_violin(cds[s.genes,], grouping = "cell_type", color_by = "cell_type")
p3 <- plot_genes_in_pseudotime(cds[s.genes,], color_by = "cell_type")
plotc <- p1|p2|p3
寻找拟时相关的基因(拟时差异基因)
Time_diff <- differentialGeneTest(cds[ordergene,], cores = 8,
fullModelFormulaStr = "~sm.ns(Pseudotime)")
Time_genes <- Time_diff %>% pull(gene_short_name) %>% as.character()
plot_pseudotime_heatmap(cds[Time_genes,], num_clusters=8, show_rownames=T, return_heatmap=T)
Reference
https://www.jianshu.com/p/5d6fd4561bc0
http://cole-trapnell-lab.github.io/monocle-release/
http://pklab.med.harvard.edu/scw2014/monocle_tutorial_oedsymbol.html
http://cole-trapnell-lab.github.io/monocle-release/tutorials/
本文由 mdnice 多平台发布