Seurat Weekly NO.0 || 开刊词
Seurat Weekly NO.1 || 到底分多少个群是合适的?!
在过去的一周里Seurat社区在github总提问数由上周的3090上升到3116,当然有同一问题反复讨论的情况,也有之前的问题还有人再问的情况,总的来说上周其在github中的issues往来邮件一共110封.本次主要和大家分享一下几个,本期的封面故事是:Randomly downsample seurat object .
在这之前,我们先讲几个Seurat的tips。读入数据,并人工生成两个分组:
library(Seurat)
pbmc <- readRDS(file = "F:\\Rstudio\\SingleR\\data\\pbmc5k.rds")
pbmc
An object of class Seurat
18792 features across 4666 samples within 1 assay
Active assay: RNA (18792 features)
3 dimensional reductions calculated: pca, tsne, umap
# pretend that cells were originally assigned to one of two replicates (we assign randomly here)
# if your cells do belong to multiple replicates, and you want to add this info to the Seurat
# object create a data frame with this information (similar to replicate.info below)
set.seed(42)
pbmc$replicate <- sample(c("rep1", "rep2"), size = ncol(pbmc), replace = TRUE)
head([email protected])
orig.ident nCount_RNA nFeature_RNA percent.mito percent.HB RNA_snn_res.0.6 seurat_clusters
AAACCCAAGCGTATGG-1 pbmc5k 13509 3498 10.659560 0 1 1
AAACCCAGTCCTACAA-1 pbmc5k 12637 3382 5.610509 0 1 1
AAACGCTAGGGCATGT-1 pbmc5k 5743 1798 10.691276 0 7 7
AAACGCTGTAGGTACG-1 pbmc5k 13107 2888 7.866026 0 9 9
AAACGCTGTGTCCGGT-1 pbmc5k 15510 3807 7.446809 0 3 3
AAACGCTGTGTGATGG-1 pbmc5k 6131 2348 9.982058 0 5 5
replicate
AAACCCAAGCGTATGG-1 rep1
AAACCCAGTCCTACAA-1 rep1
AAACGCTAGGGCATGT-1 rep1
AAACGCTGTAGGTACG-1 rep1
AAACGCTGTGTCCGGT-1 rep2
AAACGCTGTGTGATGG-1 rep2
分群和样本间标签转换
分群标签:
# Plot UMAP, coloring cells by cell type (currently stored in object@ident)
DimPlot(pbmc, reduction = "umap")

样本标签:
# How do I create a UMAP plot where cells are colored by replicate? First, store the current
# identities in a new column of meta.data called CellType
pbmc$CellType <- Idents(pbmc)
# Next, switch the identity class of all cells to reflect replicate ID
Idents(pbmc) <- "replicate"
DimPlot(pbmc, reduction = "umap")

这里注意Idents的应用,可以直接指定meta.data某一列作为[email protected].演示完了,我们再把标签换回来。
# alternately : DimPlot(pbmc, reduction = 'umap', group.by = 'replicate') you can pass the
# shape.by to label points by both replicate and cell type
# Switch back to cell type labels
Idents(pbmc) <- "CellType"
其实我们不转换ident大部分的绘图也可以用group.by来指定分群方式
DimPlot(pbmc, reduction = "umap",group.by = "replicate")
统计分群信息
# How many cells are in each cluster
table(Idents(pbmc))
0 1 2 3 4 5 6 7 8 9 10 11 12
1183 752 662 325 314 311 305 260 258 212 32 26 26
# How many cells are in each replicate?
table(pbmc$replicate)
rep1 rep2
2356 2310
prop.table(table(Idents(pbmc)))
0 1 2 3 4 5 6 7 8
0.253536219 0.161165881 0.141877411 0.069652808 0.067295328 0.066652379 0.065366481 0.055722246 0.055293613
9 10 11 12
0.045435062 0.006858123 0.005572225 0.005572225
# How does cluster membership vary by replicate?
table(Idents(pbmc), pbmc$replicate)
rep1 rep2
0 599 584
1 405 347
2 315 347
3 170 155
4 159 155
5 140 171
6 138 167
7 140 120
8 132 126
9 110 102
10 18 14
11 15 11
12 15 11
prop.table(table(Idents(pbmc), pbmc$replicate), margin = 2)
rep1 rep2
0 0.254244482 0.252813853
1 0.171901528 0.150216450
2 0.133701188 0.150216450
3 0.072156197 0.067099567
4 0.067487267 0.067099567
5 0.059422750 0.074025974
6 0.058573854 0.072294372
7 0.059422750 0.051948052
8 0.056027165 0.054545455
9 0.046689304 0.044155844
10 0.007640068 0.006060606
11 0.006366723 0.004761905
12 0.006366723 0.004761905
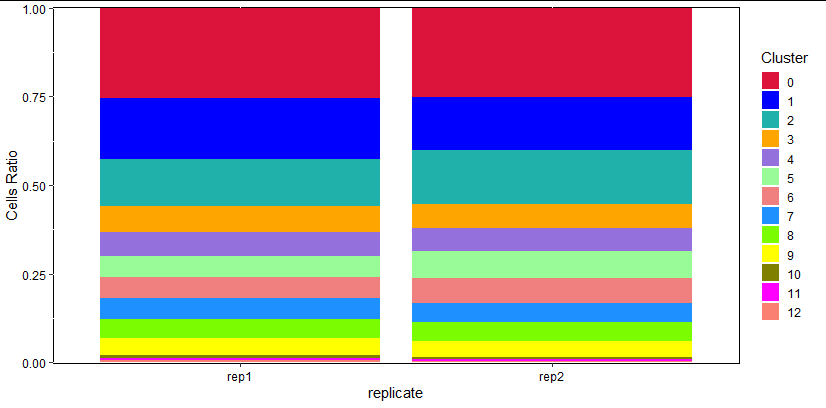
常见的频率统计图:
as.data.frame(prop.table(table(Idents(pbmc), [email protected][,"replicate"]), margin = 2))-> pdf -> td
library(tidyverse)
allcolour=c("#DC143C","#0000FF","#20B2AA","#FFA500","#9370DB","#98FB98","#F08080","#1E90FF","#7CFC00","#FFFF00","#808000","#FF00FF","#FA8072","#7B68EE","#9400D3","#800080","#A0522D","#D2B48C","#D2691E","#87CEEB","#40E0D0","#5F9EA0","#FF1493","#0000CD","#008B8B","#FFE4B5","#8A2BE2","#228B22","#E9967A","#4682B4","#32CD32","#F0E68C","#FFFFE0","#EE82EE","#FF6347","#6A5ACD","#9932CC","#8B008B","#8B4513","#DEB887")
allcolour -> colour1
plt<- ggplot(td,aes(x=td[,2],y=td[,3],fill=td[,1]))+
geom_bar(position = 'stack',stat="identity")+
labs(x="replicate",y="Cells Ratio")+
theme(panel.background=element_rect(fill='transparent', color='black'),
legend.key=element_rect(fill='transparent', color='transparent'),axis.text = element_text(color="black"))+
scale_y_continuous(expand=c(0.001,0.001))+
scale_fill_manual(values=colour1)+
guides(fill = guide_legend(keywidth = 1, keyheight = 1,ncol=1,title = 'Cluster'))
plt
取子集
Randomly downsample seurat object
github: https://github.com/satijalab/seurat/issues/3108
pbmc[, sample(colnames(pbmc), size =30, replace=F)]
An object of class Seurat
18792 features across 30 samples within 1 assay
Active assay: RNA (18792 features)
3 dimensional reductions calculated: pca, tsne, umap
当我们进入Seurat的进一步分析的时候,就会遇到这种情况:
- 取符合某种规律的
细胞出来分析 - 取符合某种规律的
基因出来分析 - 取符合某种规律的
Seurat对象 -
循环地取符合某种规律的对象 -
随机取细胞子集 - 取Seurat对象中数据存储
某一部分 - Seurat 在计算的过程中默认值的过滤,特别是基因,如:
Genes filtering prior to Normalization #3104
https://github.com/satijalab/seurat/issues/3104
missing features in SCT scale data of integrated object #3056
https://github.com/satijalab/seurat/issues/3056
有时候在做了用了相应的函数后发现基因数少了很多,其实是函数默认值卡掉了,这个是可以自己设置的。
取子的动机很简单,就是为了提出来重点分析:
- 统计某类特征
- 与其他分析结合
关于循环,我们上周讲了:Seurat Weekly NO.1 || 到底分多少个群是合适的?!
# What are the cell names of all 12 cells?
WhichCells(pbmc, idents = "12")
[1] "AAGATAGTCTTTACAC-1" "AATTCCTAGGATCACG-1" "ACCGTTCTCTTAGCAG-1" "ACCTACCGTGGACCAA-1" "AGGACTTGTAGCGAGT-1" "AGGTAGGGTACTCGAT-1" "CATTCTAAGATCGGTG-1"
[8] "CGGGTGTGTGTTACTG-1" "CTAAGTGTCCTCAGAA-1" "CTCAGGGTCAACCTTT-1" "CTCTCGACAGGTCCGT-1" "GAAATGAAGCGCACAA-1" "GAGGGTATCCTAGCTC-1" "GGATCTATCGGCTATA-1"
[15] "GGGTCTGAGCGACATG-1" "GGGTTATCATGGAGAC-1" "GTAGAGGTCAGGACAG-1" "GTAGGTTCAGGGTTGA-1" "GTTGCGGCACTTGAGT-1" "TAACACGCACTGCACG-1" "TAACTTCTCAGGTGTT-1"
[22] "TCGGGTGGTCTGTTAG-1" "TCTTTGACAAACCATC-1" "TTACAGGTCGCCTCTA-1" "TTCCAATTCCACGAAT-1" "TTCCTTCTCAACACGT-1"
特定群的count矩阵:
# How can I extract expression matrix for all ident = 12 cells (perhaps, to load into another package)
subraw.data <- as.matrix(GetAssayData(pbmc, slot = "counts")[, WhichCells(pbmc, ident = "12")])
subraw.data[1:4,1:4]
AAGATAGTCTTTACAC-1 AATTCCTAGGATCACG-1 ACCGTTCTCTTAGCAG-1 ACCTACCGTGGACCAA-1
RP11-34P13.7 0 0 0 0
FO538757.2 0 0 0 1
AP006222.2 0 1 1 0
RP4-669L17.10 0 0 0 0
Seurat 对象:
特定分组:
subset(pbmc, subset = replicate == "rep2")
An object of class Seurat
18792 features across 2310 samples within 1 assay
Active assay: RNA (18792 features)
3 dimensional reductions calculated: pca, tsne, umap
特定亚群:
# Can I create a Seurat object of just the 12 cells and 11 cells?
subset(pbmc, idents = c("12", "11"))
An object of class Seurat
18792 features across 52 samples within 1 assay
Active assay: RNA (18792 features)
3 dimensional reductions calculated: pca, tsne, umap
表达量条件:
# Can I create a Seurat object based on expression of a feature or value in object metadata?
subset(pbmc, subset = MS4A1 > 1)
An object of class Seurat
18792 features across 567 samples within 1 assay
Active assay: RNA (18792 features)
3 dimensional reductions calculated: pca, tsne, umap
排除法:
# Can I create a Seurat object of all cells except the 12 cells and 11 cells?
subset(pbmc, idents = c("12", "11"), invert = TRUE)
An object of class Seurat
18792 features across 4614 samples within 1 assay
Active assay: RNA (18792 features)
3 dimensional reductions calculated: pca, tsne, umap
计算平均表达量
# How can I calculate the average expression of all cells within a cluster?
cluster.averages <- AverageExpression(pbmc)
head(cluster.averages[["RNA"]][, 1:5])
0 1 2 3 4
RP11-34P13.7 0.006605121 0.0127983446 0.010980787 0.008408694 0.006315117
FO538757.2 0.318666146 0.5629066563 0.323559434 0.596101470 0.378227054
AP006222.2 0.076024402 0.0882946264 0.088864328 0.080678612 0.082825181
RP4-669L17.10 0.005443071 0.0081874454 0.001254630 0.005508605 0.004664880
RP5-857K21.4 0.002683901 0.0008380836 0.001704552 0.000000000 0.004602852
RP11-206L10.9 0.099380365 0.0914327250 0.099045164 0.110635758 0.109613675
返回一个平均表达的Seurat对象:
# Return this information as a Seurat object (enables downstream plotting and analysis) First,
# replace spaces with underscores '_' so ggplot2 doesn't fail
#orig.levels <- levels(pbmc)
#Idents(pbmc) <- gsub(pattern = " ", replacement = "_", x = Idents(pbmc))
#orig.levels <- gsub(pattern = " ", replacement = "_", x = orig.levels)
#levels(pbmc) <- orig.levels
cluster.averages <- AverageExpression(pbmc, return.seurat = TRUE)
cluster.averages
An object of class Seurat
18792 features across 13 samples within 1 assay
Active assay: RNA (18792 features)
head([email protected])
orig.ident nCount_RNA nFeature_RNA
0 Average 10000 16897
1 Average 10000 15744
2 Average 10000 16344
3 Average 10000 15908
4 Average 10000 14570
5 Average 10000 14523
)
啊,为什么nCount_RNA都是10000?计算平均的数据用的solt一定是data了,验证一下: ?AverageExpression果然。
# How can I calculate expression averages separately for each replicate?
cluster.averages <- AverageExpression(pbmc, return.seurat = TRUE, add.ident = "replicate")
其他
细胞相关性
CellScatter(object = pbmc_small, cell1 = 'ATAGGAGAAACAGA', cell2 = 'CATCAGGATGCACA',smooth =T)

Identifying differential expressed genes across conditions #3111
https://github.com/satijalab/seurat/issues/3111#event-3412957227
What is most likely happening is that the gene may be expressed in a small number of cells, or have outlier expression in just a few cells. This can cause it to appear to have a high expression in the 'pseudobulk', but will not return a statistically significant result when using FindMarkers. We would suggest trusting the Findmarkers output when there are discrepancies like this, but you can explore the individual genes in more detail to see what is going on. For example, please feel free to examine one of the genes that concerns you and to post a violin plot comparing the expression of any gene across conditions
数据格式转换R包:SeuratDisk
https://github.com/mojaveazure/seurat-disk
特别是Seurat and Scanpy.之间的对象传输。