24《Protein Actions Principles and Modeling》-《蛋白质作用原理和建模》中文分享
《Protein Actions Principles and Modeling》-《蛋白质作用原理和建模》
本人能力有限,如果错误欢迎批评指正。
第六章:The principles of protein folding kinetics
(蛋白质折叠动力学的原理)
-Levinthal悖论促进蛋白质折叠机制研究
在本章,我们将会了解蛋白质折叠/展开的速率和途径。我们所说的路线是指当蛋白质折叠时发生的构象变化的时间过程。我们会了解到在折叠过程中,蛋白质在什么阶段获得了其二级结构、其氢键、其疏水核心和其紧密的侧链堆积。那么是否这里会存在一种比较普遍的机制可以用来解释折叠/展开过程中的共性呢?这些过程是否与某些因素如温度或者变性剂有关呢?蛋白质如何快速发现自己适合的折叠结构并且同时从展开状态开始折叠-有时候该过程只需要几毫秒?什么会是折叠速度,其中的物理依据是什么?
为什么蛋白质折叠动力学那么重要?因为折叠动力学对于蛋白质科学的历史发展的重点。在1968年,Cyrus Levinthal提出了Levinthal悖论。蛋白质存在一个大海捞针的问题。蛋白质的构想空间是个非常大的空间-大海,一个非折叠状态的蛋白质必须找到折叠状态的天然构象-针。如果我们假设每隔蛋白单元都具有z=3的8个构象,那么当一个蛋白的氨基酸有100个的时候,理论上它的构象为zN ≈ 1050–1090。那么Levinthal悖论得到问题在于蛋白质如何是在这么大的搜索空间中快速找到那个天然的结构-过程只需要微秒-毫秒。
Levinthal悖论引发了以下的想法:如果我们了解折叠的途径,那么我们也许可以或者一个折叠规律/机制-不同的蛋白质在寻找其天然构象的时候,哪些构想会被抛弃/考虑。也许了解折叠中间产物(展开-折叠过程中的中间产物)可以给我们一些线索去了解哪些已知蛋白的构象是无需考虑的。当我们已知蛋白质序列的时候,我们可以利用这些知识来预测出蛋白质的结构。折叠动力学的知识同时也有利了解其他过程的动力学:配体结合,构象的变构变化,和机制作用。
-Mass-Action 模型可以进行折叠速率实验的捕捉
我们从最基础的折叠动力学实验开始学习。先从稀释的蛋白质溶液(变性条件的控制下)开始。我们可以通过急剧改变变性剂的浓度,Ph,温度来使该溶液的蛋白质转变为折叠状态。然后我们观察随着时间的变化光学特性(圆二色性(CD)或荧光,y (t))的变化。我们可以监测试管中折叠蛋白数量增加的速率。这种类型的实验通常被称为体外重折叠(in vitro refolding),因为它发生在试管中,而不是在细胞内,与最初未展开的蛋白质一起进行。我们也可以反向运行实验,通过将蛋白质溶液从折叠条件跳到展开条件来展开天然蛋白质。实验使用的是稀释的蛋白质溶液,可以避免蛋白质聚集。折叠和展开过程通常用一个或多个时间指数函数很好地描述(图 6.1):
![]()
速率系数k1和k2以及振幅A1和A2可以你深入了解不同条件下的折叠过程。

图6.1 折叠和展开通常遵循一个或两个指数衰减。通过(A)溶菌酶和(B)细胞色素c的光吸光度,以及(C)核糖核酸酶a的荧光反应,天然或变性蛋白的数量随时间的变化。线性是这些对数图上的一条直线。图显示了溶菌酶的双态折叠和展开动力学;细胞色素c的三态折叠和展开;以及核糖核酸酶a的双态折叠和线性展开的结合。
在本章中,我们将描述从宏观和微观模型中获得的知识。在宏观模型中,我们的目标是选择适合我们数据的Mass-Action 模型。Mass-Action模型是一组选定的动力学状态如D、I和N,以及连接这些状态的假定动力学方案(图6.2)。宏观建模只是用简洁的语言来表达实验结果的途径。微观模型旨在捕捉潜在的物理学——分子的构象和能量如何影响蛋白质折叠速率和路径。微观模型的目的是对折叠过程中的时间,温度、变性剂、pH或氨基酸序列对折叠过程的影响,给出一个结构和物理的解释。首先,让我们从宏观模型开始吧。

图6. 2 折叠的Mass-Action模型。(A)双态模型。(B)在通路上的中间状态。(C)通路中间体。(D)一个具有多个中间体的线性模型。
小蛋白质通过双态动力学快速折叠
一个典型的小的单结构域蛋白质只在一个单指数弛豫过程中折叠或展开,如图6.1A所示的溶菌酶。然界中涉及单指数动力学的过程据说遵循双态动力学,因为你需要解释它们的Mass-Action模型只需要两种状态,在这种情况下是D(展开)和N(折叠):

其中kf和ku分别为折叠和展开速率系数。这一过程的时间依赖性可以描述为:
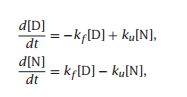
其中,[D]和[N]分别是展开状态和折叠状态的时间依赖性浓度。如果选择归一化单位使[D] + [N] = 1,那么[D]和[N]是时间依赖的概率或总体-pD (t)和pN (t),值范围从0到1。有不同的方法来解决方程6.3。最简单的方法是用pD (t) = 1−pN (t)求解pN (t)。然而,这种策略主要局限于双态动力学。因为我们想探索适用于不同类型动力学的更广泛的模型,所以我们使用了一种更常用的方法,称为主方程。在这种方法中,在列向量中表示状态概率:

而速率系数为:
![]()
概率的时间依赖性为:

那么在两个双态转变中,折叠的结构的比例为

而展开状态的概率为 pD(t) = 1 − pN(t)。平衡状态的折叠分子比例为
![]()
方程6.7表明,如果在时间t = 0跳转环境,我们将观察到一个单指数衰减到带有一个速率系数的平衡状态:
![]()
在一个限制中,我们可以从时间t = 0开始,所有分子展开,pN (0) = 0,并应用强折叠条件,使ku→0。然后,折叠结构的概率将随时间呈指数增长,速率系数为kf
![]()
在相反的限制下,我们可以从时间t = 0开始,所有分子完全折叠,pN (0) = 1,并应用强变性条件,使kf→0。然后,折叠结构将以指数级消失:
![]()
这就是如何使用公式6.7-调整参数kf和ku,以达到方程6.7与实验数据的最佳拟合。如果数据比单指数动态更复杂,那么我们将尝试一个不同的模型,通常是一个具有中间状态的模型。前面描述的双态动力学对于小的蛋白质通常是足够的。但我们应该如何解释这种指数级的时间依赖性?这是否意味着试管中的所有蛋白质都是同步的,例如,每个分子形成第一个螺旋,同时其他蛋白质分子形成第一个螺旋结构?还是这意味着每个蛋白质本身折叠得非常快,没有与其他蛋白质分子同步,指数衰减只是反映了在给定时间内折叠或展开?后者才是对的。速率系数kf和ku是单位时间内任何特定蛋白质分子转化的概率。在任何给定的时间内,转化为N的分子的数量与当时在试管中仍未转化的D分子的数量成正比。溶液中的D分子越少,在单位时间内发生的D→N转换就越少。这是一种浓度效应。这是蛋白质折叠或展开的指数时间过程的基础。方框6.1展示了如何使用这种解释从计算机模拟中确定折叠速率。
| Box 6.1 通过多个短的计算机模拟来估计一个蛋白质的折叠速率 |
| 假设我们运行一个计算机折叠模拟来确定折叠速率系数kf。(这些模拟将在第10章中进行讨论。)我们既可以跑一些长轨迹,也可以跑许多短轨迹。我们可以从一些未展开的构象开始模拟,然后在折叠条件下运行蛋白质的长轨迹。计算机轨迹应该运行至少几个时间的倍数,τf = 1/kf,折叠速率系数的倒数。否则,您将没有足够的数据来准确地拟合松弛曲线的基线。例如,如果真正的折叠时间为1 μs,则至少需要5-10μs的模拟时间。但在计算机上运行长时间的运行轨迹需要很长时间。相反,如果你有并行的计算机,你可以运行许多短的轨迹并收集统计数据。V Pande在一个名为折叠@home的计算机资源上展示了这一点。式6.10表明,对于短时间(t<<τf),从变性状态开始,可以将原生状态的时间变总体近似为
重新排列方程6.12给出
通过这种方式,可以从许多短轨迹中估计kf,而不是更少的长轨迹。只需计算一下你所看到的折叠情况的分数。这种策略很有用,因为在许多较短的运行中通常比在许多较短的运行中更容易获取计算机资源。而较长的轨迹并不一定能提供足够的构象空间采样。 |
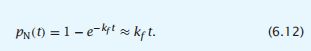

小的球状蛋白质(小于大约100-150个氨基酸)通常通过双态动力学折叠。值得注意的是,蛋白质折叠具有如此简单的动力学,因为它们的异相和复杂的分子结构。我们在下面表明,任何具有单指数动力学的过程都可以被描述为具有一个速率限制的步骤,或一个过渡态。
-------------------------------------------
欢迎点赞收藏转发!
下次见!