- 用AI实现“抢券自由”:手把手教你打造智能抢单机器人
目录一、手速不够?抢券党的真实困境二、技术揭秘:RPA+AI如何成为“抢券外挂”1.什么是RPA(机器人流程自动化)?2.AI工作流的降维打击三、实战教学:20行代码打造AI抢券机器人1.工具准备2.智能脚本核心代码四、高阶技巧:让机器人更“聪明”的3个秘密1.视觉识别加持2.多账号并行操作3.智能避坑策略五、技术延伸:RPA+AI还能做什么?六、避坑指南:新手常见问题解答一、手速不够?抢券党的真
- 支付系统对接与订单生命周期全流程解析:企业级 SaaS 与在线服务场景的实战落地指南
观熵
企业级SaaS架构与工程实战全流程java网络服务器Saas
支付系统对接与订单生命周期全流程解析:企业级SaaS与在线服务场景的实战落地指南关键词支付系统接入、订单生命周期、支付平台对接、支付状态机、订单幂等性处理、支付成功回调、自动对账、退款处理、Webhook安全、支付异常监控摘要在构建具备商业化能力的SaaS产品或在线服务平台时,支付系统的接入与订单生命周期管理是支撑订阅、计费与收入闭环的关键环节。本篇文章将系统性解析企业如何对接主流支付平台(如St
- 租户订阅、套餐切换与服务启停全流程设计:SaaS 计费引擎与运营控制体系实战解析
观熵
企业级SaaS架构与工程实战全流程SaaS架构
租户订阅、套餐切换与服务启停全流程设计:SaaS计费引擎与运营控制体系实战解析关键词多租户订阅系统、SaaS套餐管理、服务启停机制、计费周期控制、套餐额度配置、权限策略切换、租户状态机、运营控制后台、资源配额调节、合约期与续订策略摘要在企业级SaaS系统中,租户订阅与套餐管理机制不仅决定平台的盈利模型,更直接关系到权限控制、资源分配、服务启停等系统级行为。传统CRM或权限表驱动的权限体系难以支撑“
- 多探头分布式雷达测流系统解决方案概述
一、雷达测流的方案背景近年来,雷达测流作为一种新的测量方式,正在不断被引进和使用,其旨在解决传统测量方式无法解决的问题或难题。传统的测量方式,如直接接触式测流,受到多种因素的影响,如水中含沙量、漂浮物、气候等,导致测量结果不准确。而雷达测流设备则以非接触方法测量水体表面流速,不受水中含沙量、漂浮物、气候等因素影响,适用于一般河流、污水流速等测量。此外,雷达测流设备还特别适用于夹带污物的排水、高洪和
- 【领码思考】ESG画卷里的项目管理新篇:AI赋能下的绿色智造之路
领码科技
央国企理念篇AI应用人工智能ESG项目管理AI赋能绿色转型可持续发展
摘要ESG(环境、社会、治理)理念正悄然融入项目管理的每个细胞,成为驱动项目成功的新引擎。本文聚焦ESG如何与项目管理深度融合,立体呈现各阶段ESG应用场景,围绕AI与数字化工具的协同赋能,解析项目经理如何在绿色转型中实现角色跃迁。通过流程图与表格精炼框架,强化理论指导与实践操作,并结合当下热点新技术,旨在为项目团队和企业管理层提供清晰可落地的全周期ESG实施路径,开启项目管理可持续发展的智慧新纪
- 基于评估方法论评估一个大模型的准确度
尤物程序猿
自动化运维
评估标准先来说说什么是大模型的一个准确度,指其输出结果与真实值或期望值之间的符合程度,但在不同任务和场景下具体定义和评估方式存在显著差异。要评估一个大模型还得考虑到评估哪些方面呢?以下是大概的几个方向任务类型准确度定义分类任务预测类别与真实标签的一致性生成任务生成内容的真实性/流畅性/相关性问答任务答案的事实正确性和完整性多模态任务跨模态对齐能力(如图文匹配)除了以上几个方面还需要考虑表面匹配:字
- 智能电动汽车 --- 车辆网关路由缓存
汽车电子实验室
电子电器架构—车载网关缓存ZEVonUDS-J1979OEM怎么掌握软件开发能力车载通信网络槪述HPC软件架构汽车
我是穿拖鞋的汉子,魔都中坚持长期主义的汽车电子工程师。老规矩,分享一段喜欢的文字,避免自己成为高知识低文化的工程师:简单,单纯,喜欢独处,独来独往,不易合同频过着接地气的生活,除了生存温饱问题之外,没有什么过多的欲望,表面看起来很高冷,内心热情,如果你身边有这样灵性的人,一定要好好珍惜他们眼中有神有光,干净,给人感觉很舒服,有超强的感知能力有形的无形的感知力很强,能感知人的内心变化喜欢独处,好静,
- 物联网零售领域AI算力网络与通信的应用探索
AI算力网络与通信
物联网零售人工智能ai
物联网零售领域AI算力网络与通信的应用探索关键词:物联网、零售领域、AI算力网络、通信、应用探索摘要:本文聚焦于物联网零售领域,深入探讨了AI算力网络与通信的应用。首先介绍了相关背景,包括目的、预期读者等。接着对核心概念进行解释,阐述它们之间的关系并给出原理架构示意图和流程图。然后详细讲解核心算法原理、数学模型与公式,通过项目实战展示代码案例及解读。还介绍了实际应用场景、推荐相关工具资源,分析未来
- Golang 与 Kafka 的协同:优化消息处理流程
Golang编程笔记
golangkafkalinqai
Golang与Kafka的协同:优化消息处理流程关键词:Golang、Kafka、消息队列、并发处理、性能优化、消费者组、异步通信摘要:本文将带你探索如何用Golang的“轻量级并发魔法”与Kafka的“高吞吐量消息引擎”协同工作,优化消息处理流程。我们会从基础概念到实战案例,用“快递站分包裹”“餐厅传菜”等生活场景类比,一步步拆解技术细节,最终掌握如何让这对“黄金组合”高效处理百万级消息。背景介
- 探索AI人工智能领域多智能体系统的技术原理
AI大模型应用之禅
人工智能网络ai
探索AI人工智能领域多智能体系统的技术原理关键词:AI人工智能、多智能体系统、技术原理、智能体交互、分布式计算摘要:本文深入探索了AI人工智能领域多智能体系统的技术原理。首先介绍了多智能体系统的背景,包括其目的、预期读者、文档结构和相关术语。接着阐述了多智能体系统的核心概念与联系,通过文本示意图和Mermaid流程图进行清晰展示。详细讲解了核心算法原理,结合Python源代码进行说明,并给出了相关
- 【科研写作自动化工具】如何用AI技术组合(大模型+多Agent+自动化)打造一个“智能论文生产线”,把枯燥的写作流程变成自动化
n8n是一款开源的工作流自动化工具,类似于Zapier或Make(原Integromat),但更注重灵活性和开发者友好性。在课程文件中提到的n8n自动化流水线主要用于科研写作的自动化流程集成,以下是详细解释:n8n的核心功能可视化工作流设计:通过拖拽节点(Nodes)连接不同工具和服务,无需编写复杂代码即可搭建自动化流程。多平台集成:支持连接文献数据库(如PubMed、arXiv)、AI模型(如O
- GitHub Actions × AWS 集成终极指南:从零构建安全高效的CI/CD流水线
ivwdcwso
运维与云原生githubaws安全GitHubActionsDevOpsCI/CD
引言:云原生时代的自动化革命在DevOps实践中,GitHubActions与AWS的深度集成已成为现代应用交付的黄金标准。这种组合让开发者能够:✅实现端到端自动化:从代码提交到生产部署的全流程自动化内置企业级安全:通过OIDC消除密钥泄露风险优化资源成本:按执行分钟计费,无闲置资源浪费加速迭代速度:将部署时间从小时级缩短至分钟级本文将全面解析最佳实践、安全策略和高级技巧,助您构建工业级CI/CD
- 【剪裁Patch】已标注的WSI剪裁Patch的处理流程(以QuPath软件得到的标注信息为例)
X CODE
WSI病理图像QuPathPython
1.整体处理思路整体处理流程如图所示,概括来说就是:根据标注信息将WSI区分为肿瘤区域和正常区域,对这个区域进行采样裁剪得到具有Patch级别标签的Patch。当然,这里的Patch标签是根据标注信息决定的,如果标注的是癌症亚型信息,那么也可以将不同亚型的Patch区分出来。那么下面就对每个步骤进行介绍以及提供具体的Python代码。2.详细步骤(提供代码)2.0标注文件示例以下是用QuPath软
- Python 领域 Conda 的集群环境部署经验
Python领域Conda的集群环境部署经验关键词:Conda、Python环境管理、集群部署、环境复制、依赖管理、虚拟环境、Anaconda摘要:本文深入探讨了在集群环境中使用Conda进行Python环境部署的最佳实践。我们将从Conda的核心概念出发,详细讲解环境创建、依赖管理、环境复制和集群部署的全流程。文章包含实际案例、性能优化技巧和常见问题解决方案,旨在帮助读者掌握高效、可靠的Pyth
- 【网络通信安全】深入解析 OSPF 协议:从概念到 eNSP 实战配置(附完整代码与排错指南)
不羁。。
网络通信安全智能路由器网络
目录一、OSPF协议核心概念:为什么它是企业网络的“神经网络”?1.协议本质与设计目标2.核心组件与工作原理(1)链路状态数据库(LSDB)(2)区域划分原则(3)路由器角色二、实验环境搭建:3台路由器构建跨区域OSPF网络1.网络拓扑图2.设备与IP规划表三、逐设备配置详解:从接口到OSPF进程的全流程操作1.基础配置:接口IP与设备命名(以R1为例)2.OSPF进程配置:区域划分与网络宣告(1
- Dockerfile 设定镜像时区
半吊子游戏开发者
我整理的一些关于【Docker】的项目学习资料(附讲解~~)和大家一起分享、学习一下:https://d.51cto.com/f2PFnNDockerfile设定镜像时区指南在Docker中,镜像的时区配置常常被忽视,但这对于确保应用在运行时的时间正确性至关重要。在这篇文章中,我将指导你如何通过Dockerfile设定镜像的时区,并提供一个清晰的操作流程。一、步骤流程我们将整个过程分为几个步骤,以
- 【AIDD药物研发】张载熙-生成式AI4药物发现
静静喜欢大白
医疗影像人工智能AIDD药物研究药物生成生成
目录1、简介2、生成式AI用于基于结构式的药物发现背景生成用于靶标结合的类药小分子功能性蛋白质的生成与优化其他新的药物形式及生物安全/安全性小结3、参考4、补充学习资料1、简介最近需要简单了解喜爱AIDD流程以及相关进展调研,看到zaixizhang正在做相关研究,进行下面的学习记录张载熙中国科学技术大学计算机科学与技术学院2021级博士生(导师刘淇教授),认知智能全国重点实验成员,本科毕业于中国
- python之数据库操作
婵婵子~
python数据库python
python操作sqlserver数据库python标准数据库接口为pythonDB-API,具体可参考:https://wiki.python.org/moin/DatabaseInterfacesPython的DB-API,为大多数的数据库实现了接口,使用它连接各数据库后,就可以用相同的方式操作各数据库。PythonDB-API使用流程:引入API模块。获取与数据库的连接。执行SQL语句和存储
- AI大模型如何重塑软件开发流程?
真实的菜
活动人工智能
AI大模型如何重塑软件开发流程?文章摘要随着ChatGPT、Claude等AI大模型的快速发展,软件开发行业正经历着前所未有的变革。本文深入探讨了AI技术如何重塑传统的软件开发流程,分析了开发者角色的转变,并提供了拥抱AI时代的实践指南。核心观点AI大模型将开发者角色从"编码者"转变为"设计师"需求分析、代码生成、测试等环节将实现智能化新技能需求:AI工具使用、提示工程、跨领域整合未来趋势:低代码
- 深入解析u-boot-1.1.6源码与应用
kdbshi
本文还有配套的精品资源,点击获取简介:u-boot-1.1.6是一款重要的开源引导加载程序,广泛应用于嵌入式系统。本文对u-boot-1.1.6版本源码进行深入剖析,帮助读者理解其工作原理、主要功能及关键模块。内容涉及u-boot简介、源码结构、启动流程、关键功能、学习与调试方法,并总结了其在嵌入式系统中的重要性。本文旨在通过细致研究源码,提升开发者对嵌入式系统的理解与应用能力。1.u-boot概
- 动手实践OpenHands系列学习笔记11:现代开发流程
笔记11:现代开发流程一、引言现代软件开发流程是确保高质量代码交付和团队协作的关键基础。随着软件开发复杂度的增加,自动化工具链和规范化流程变得尤为重要。本笔记将探讨CI/CD管道设计原理,分析OpenHands项目的开发流程,并通过实践搭建一个简化版的OpenHands开发环境。二、CI/CD管道设计理论2.1持续集成(CI)基本概念定义:频繁地将代码集成到主分支,并自动化验证每次集成核心原则:频
- Badoo×亚矩云手机:社交约会革命的“云端心跳加速剂“
云云321
智能手机网络游戏大数据矩阵
——当全球最大陌生人社交平台遇上云端算力,破解"颜值即正义"困局,重塑真实、高效、安全的下一代社交体验作为月活超4亿的全球陌生人社交巨头,Badoo以"附近的人+动态分享"模式,帮助用户跨越地理与社交圈层建立连接。然而,随着Z世代对"真实社交、深度互动、隐私安全"的需求爆发,Badoo正面临三大核心挑战:颜值内卷与虚假人设:过度依赖照片/视频的展示方式,导致"照骗"泛滥,用户匹配后见面破灭率高达6
- Java的神奇绘图功能:画一条直线
一、背景引入第一篇介绍了如何设置一个简单的登录界面,今天就来讲讲界面JFrame的其他功能:绘图,但作为向递归分形的过渡内容,我们今天不需要画出多复杂多精美的图案,只需要在界面上能够画出一条简单的直线即可。二、问题思考1.摆在眼前的问题与资源需要解决的问题:如何实现画一条直线?可以解决问题的资源:有关Java的一些基础知识和简单的界面基础2.怎么画一条直线(1)猜想:画一条直线的可能流程首先是画一
- Jenkins集成GitHub实现自动化打标签实战指南
ivwdcwso
运维与云原生jenkinsgithub自动化CI/CDdevops
本文将详细介绍如何使用Jenkins与GitHubAPI集成,实现自动化打标签的完整流程。以下是完整的Python脚本和详细解析。完整Python脚本#!/root/miniconda3/bin/pythonimportjsonimportboto3importosimportpytzimportargparsefromdatetimeimportdatetimefromgithubimportG
- 超详细yolov8/11-segment实例分割全流程概述:配置环境、数据标注、训练、验证/预测、onnx部署(c++/python)详解
因为yolo的检测/分割/姿态/旋转/分类模型的环境配置、训练、推理预测等命令非常类似,这里不再详细叙述,主要参考**【YOLOv8/11-detect目标检测全流程教程】**,下面有相关链接,这里主要针对数据标注、格式转换、模型部署等不同细节部分;【YOLOv8/11-detect目标检测全流程教程】超详细yolo8/11-detect目标检测全流程概述:配置环境、数据标注、训练、验证/预测、o
- LabVIEW MathScript薄板热流模拟
LabVIEW开发
LabVIEW参考程序LabVIEW知识LabVIEW知识LabVIEW程序LabVIEW功能labview
热流模拟是热设计关键环节,传统工具精准但开发周期长,本VI利用LabVIEW优势,面向工程师快速验证需求,在初步方案迭代、教学演示等场景更具效率,为热分析提供轻量化替代路径,后续可结合专业工具,先通过本VI快速定性分析,再用传统工具精准求解,提升研发流程效率。此VI用于模拟单点热源下薄板的热流,求解带周期边界条件的椭圆型偏微分方程,借助LabVIEWMathScriptNode实现自定义函数,结合
- PHP接单涨薪系列(九)之计算机视觉实战:PHP+Stable Diffusion接单指南(2025高溢价秘籍)
攻城狮凌霄
PHPPHP接单涨薪AIphp计算机视觉stablediffusion
案例场景某电商公司使用本方案后,产品图制作成本降低90%,广告转化率提升35%,单月节省设计费用超¥80,000。本文将彻底解密如何用PHP+AI视觉技术接取高单价设计外包,让你在竞争激烈的市场中脱颖而出!一、视觉设计市场的AI革命1.1传统设计vsAI设计设计任务传统流程AI流程需求沟通初稿设计反复修改最终交付AI生成微调即时交付2025年设计市场数据对比:指标传统设计AI设计提升幅度单图制作时
- Python(28)Python循环语句指南:从语法糖到CPython字节码的底层探秘
一个天蝎座白勺程序猿
Python爬虫入门到高阶实战python开发语言
目录引言一、推导式家族全解析1.1基础语法对比1.2性能对比测试二、CPython实现揭秘2.1字节码层面的秘密2.2临时变量机制三、高级特性实现3.1嵌套推导式优化3.2条件表达式处理四、性能优化指南4.1内存使用对比4.2执行时间优化技巧五、最佳实践建议六、总结Python爬虫相关文章(推荐)引言在Python编程中,循环语句是控制流程的核心工具。传统for循环虽然直观,但在处理大数据时往往面
- 【多线程】线程的引入,创建线程的方式,设置线程名字、获取名字,线程优先级priority,加入休眠的方法,,后台线程,礼让线程,Join,中断线程,某电影院,共有100张票线程流程图,3售票窗口,
心盲i1
Java基础多线程java
多线程1.线程的引入进程:正在运行的程序,是系统进行资源分配和调用的独立单位。每一个进程都有它自己的内存空间和资源。线程:是进程的单个顺序控制流,或者说就是一个单独执行的路径一个进程如果只有一条执行路径,称之为单线程一个进程如果有多条执行路径,称之为多线程线程是包含在进程中。举例:扫雷,360杀毒软件,百度网盘了解三个关键词:1、串行,指的是一个程序中所有的任务都是按照先后顺序执行的,在前一个任务
- 国内中小制造业“内卷”困局六大问题:盟接之桥的六大建议
盟接之桥
制造人工智能大数据服务器运维数据可视化
近年来,中国制造业正面临前所未有的“内卷”压力。企业不仅要应对不断压缩的利润空间,还需在复杂的供应链、生产流程和客户需求之间艰难平衡。本文基于多位制造业老板的实际反馈,梳理当前行业的主要痛点,并结合“盟接之桥”的专业建议,提出切实可行的破局路径。一、当前制造业面临的六大核心问题账期压力加剧现金流紧张大客户普遍要求3个月账期,甚至6个月银行承兑汇票结算。导致企业资金链长期处于高压状态,尤其对中小型企
- apache ftpserver-CentOS config
gengzg
apache
<server xmlns="http://mina.apache.org/ftpserver/spring/v1"
xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance"
xsi:schemaLocation="
http://mina.apache.o
- 优化MySQL数据库性能的八种方法
AILIKES
sqlmysql
1、选取最适用的字段属性 MySQL可以很好的支持大数据量的存取,但是一般说来,数据库中的表越小,在它上面执行的查询也就会越快。因此,在创建表的时候,为了获得更好的 性能,我们可以将表中字段的宽度设得尽可能小。例如,在定义邮政编码这个字段时,如果将其设置为CHAR(255),显然给数据库增加了不必要的空间,甚至使用VARCHAR这种类型也是多余的,因为CHAR(6)就可以很
- JeeSite 企业信息化快速开发平台
Kai_Ge
JeeSite
JeeSite 企业信息化快速开发平台
平台简介
JeeSite是基于多个优秀的开源项目,高度整合封装而成的高效,高性能,强安全性的开源Java EE快速开发平台。
JeeSite本身是以Spring Framework为核心容器,Spring MVC为模型视图控制器,MyBatis为数据访问层, Apache Shiro为权限授权层,Ehcahe对常用数据进行缓存,Activit为工作流
- 通过Spring Mail Api发送邮件
120153216
邮件main
原文地址:http://www.open-open.com/lib/view/open1346857871615.html
使用Java Mail API来发送邮件也很容易实现,但是最近公司一个同事封装的邮件API实在让我无法接受,于是便打算改用Spring Mail API来发送邮件,顺便记录下这篇文章。 【Spring Mail API】
Spring Mail API都在org.spri
- Pysvn 程序员使用指南
2002wmj
SVN
源文件:http://ju.outofmemory.cn/entry/35762
这是一篇关于pysvn模块的指南.
完整和详细的API请参考 http://pysvn.tigris.org/docs/pysvn_prog_ref.html.
pysvn是操作Subversion版本控制的Python接口模块. 这个API接口可以管理一个工作副本, 查询档案库, 和同步两个.
该
- 在SQLSERVER中查找被阻塞和正在被阻塞的SQL
357029540
SQL Server
SELECT R.session_id AS BlockedSessionID ,
S.session_id AS BlockingSessionID ,
Q1.text AS Block
- Intent 常用的用法备忘
7454103
.netandroidGoogleBlogF#
Intent
应该算是Android中特有的东西。你可以在Intent中指定程序 要执行的动作(比如:view,edit,dial),以及程序执行到该动作时所需要的资料 。都指定好后,只要调用startActivity(),Android系统 会自动寻找最符合你指定要求的应用 程序,并执行该程序。
下面列出几种Intent 的用法
显示网页:
- Spring定时器时间配置
adminjun
spring时间配置定时器
红圈中的值由6个数字组成,中间用空格分隔。第一个数字表示定时任务执行时间的秒,第二个数字表示分钟,第三个数字表示小时,后面三个数字表示日,月,年,< xmlnamespace prefix ="o" ns ="urn:schemas-microsoft-com:office:office" />
测试的时候,由于是每天定时执行,所以后面三个数
- POJ 2421 Constructing Roads 最小生成树
aijuans
最小生成树
来源:http://poj.org/problem?id=2421
题意:还是给你n个点,然后求最小生成树。特殊之处在于有一些点之间已经连上了边。
思路:对于已经有边的点,特殊标记一下,加边的时候把这些边的权值赋值为0即可。这样就可以既保证这些边一定存在,又保证了所求的结果正确。
代码:
#include <iostream>
#include <cstdio>
- 重构笔记——提取方法(Extract Method)
ayaoxinchao
java重构提炼函数局部变量提取方法
提取方法(Extract Method)是最常用的重构手法之一。当看到一个方法过长或者方法很难让人理解其意图的时候,这时候就可以用提取方法这种重构手法。
下面是我学习这个重构手法的笔记:
提取方法看起来好像仅仅是将被提取方法中的一段代码,放到目标方法中。其实,当方法足够复杂的时候,提取方法也会变得复杂。当然,如果提取方法这种重构手法无法进行时,就可能需要选择其他
- 为UILabel添加点击事件
bewithme
UILabel
默认情况下UILabel是不支持点击事件的,网上查了查居然没有一个是完整的答案,现在我提供一个完整的代码。
UILabel *l = [[UILabel alloc] initWithFrame:CGRectMake(60, 0, listV.frame.size.width - 60, listV.frame.size.height)]
- NoSQL数据库之Redis数据库管理(PHP-REDIS实例)
bijian1013
redis数据库NoSQL
一.redis.php
<?php
//实例化
$redis = new Redis();
//连接服务器
$redis->connect("localhost");
//授权
$redis->auth("lamplijie");
//相关操
- SecureCRT使用备注
bingyingao
secureCRT每页行数
SecureCRT日志和卷屏行数设置
一、使用securecrt时,设置自动日志记录功能。
1、在C:\Program Files\SecureCRT\下新建一个文件夹(也就是你的CRT可执行文件的路径),命名为Logs;
2、点击Options -> Global Options -> Default Session -> Edite Default Sett
- 【Scala九】Scala核心三:泛型
bit1129
scala
泛型类
package spark.examples.scala.generics
class GenericClass[K, V](val k: K, val v: V) {
def print() {
println(k + "," + v)
}
}
object GenericClass {
def main(args: Arr
- 素数与音乐
bookjovi
素数数学haskell
由于一直在看haskell,不可避免的接触到了很多数学知识,其中数论最多,如素数,斐波那契数列等,很多在学生时代无法理解的数学现在似乎也能领悟到那么一点。
闲暇之余,从图书馆找了<<The music of primes>>和<<世界数学通史>>读了几遍。其中素数的音乐这本书与软件界熟知的&l
- Java-Collections Framework学习与总结-IdentityHashMap
BrokenDreams
Collections
这篇总结一下java.util.IdentityHashMap。从类名上可以猜到,这个类本质应该还是一个散列表,只是前面有Identity修饰,是一种特殊的HashMap。
简单的说,IdentityHashMap和HashM
- 读《研磨设计模式》-代码笔记-享元模式-Flyweight
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.util.ArrayList;
import java.util.Collection;
import java.util.HashMap;
import java.util.List;
import java
- PS人像润饰&调色教程集锦
cherishLC
PS
1、仿制图章沿轮廓润饰——柔化图像,凸显轮廓
http://www.howzhi.com/course/retouching/
新建一个透明图层,使用仿制图章不断Alt+鼠标左键选点,设置透明度为21%,大小为修饰区域的1/3左右(比如胳膊宽度的1/3),再沿纹理方向(比如胳膊方向)进行修饰。
所有修饰完成后,对该润饰图层添加噪声,噪声大小应该和
- 更新多个字段的UPDATE语句
crabdave
update
更新多个字段的UPDATE语句
update tableA a
set (a.v1, a.v2, a.v3, a.v4) = --使用括号确定更新的字段范围
- hive实例讲解实现in和not in子句
daizj
hivenot inin
本文转自:http://www.cnblogs.com/ggjucheng/archive/2013/01/03/2842855.html
当前hive不支持 in或not in 中包含查询子句的语法,所以只能通过left join实现。
假设有一个登陆表login(当天登陆记录,只有一个uid),和一个用户注册表regusers(当天注册用户,字段只有一个uid),这两个表都包含
- 一道24点的10+种非人类解法(2,3,10,10)
dsjt
算法
这是人类算24点的方法?!!!
事件缘由:今天晚上突然看到一条24点状态,当时惊为天人,这NM叫人啊?以下是那条状态
朱明西 : 24点,算2 3 10 10,我LX炮狗等面对四张牌痛不欲生,结果跑跑同学扫了一眼说,算出来了,2的10次方减10的3次方。。我草这是人类的算24点啊。。
然后么。。。我就在深夜很得瑟的问室友求室友算
刚出完题,文哥的暴走之旅开始了
5秒后
- 关于YII的菜单插件 CMenu和面包末breadcrumbs路径管理插件的一些使用问题
dcj3sjt126com
yiiframework
在使用 YIi的路径管理工具时,发现了一个问题。 <?php
- 对象与关系之间的矛盾:“阻抗失配”效应[转]
come_for_dream
对象
概述
“阻抗失配”这一词组通常用来描述面向对象应用向传统的关系数据库(RDBMS)存放数据时所遇到的数据表述不一致问题。C++程序员已经被这个问题困扰了好多年,而现在的Java程序员和其它面向对象开发人员也对这个问题深感头痛。
“阻抗失配”产生的原因是因为对象模型与关系模型之间缺乏固有的亲合力。“阻抗失配”所带来的问题包括:类的层次关系必须绑定为关系模式(将对象
- 学习编程那点事
gcq511120594
编程互联网
一年前的夏天,我还在纠结要不要改行,要不要去学php?能学到真本事吗?改行能成功吗?太多的问题,我终于不顾一切,下定决心,辞去了工作,来到传说中的帝都。老师给的乘车方式还算有效,很顺利的就到了学校,赶巧了,正好学校搬到了新校区。先安顿了下来,过了个轻松的周末,第一次到帝都,逛逛吧!
接下来的周一,是我噩梦的开始,学习内容对我这个零基础的人来说,除了勉强完成老师布置的作业外,我已经没有时间和精力去
- Reverse Linked List II
hcx2013
list
Reverse a linked list from position m to n. Do it in-place and in one-pass.
For example:Given 1->2->3->4->5->NULL, m = 2 and n = 4,
return
- Spring4.1新特性——页面自动化测试框架Spring MVC Test HtmlUnit简介
jinnianshilongnian
spring 4.1
目录
Spring4.1新特性——综述
Spring4.1新特性——Spring核心部分及其他
Spring4.1新特性——Spring缓存框架增强
Spring4.1新特性——异步调用和事件机制的异常处理
Spring4.1新特性——数据库集成测试脚本初始化
Spring4.1新特性——Spring MVC增强
Spring4.1新特性——页面自动化测试框架Spring MVC T
- Hadoop集群工具distcp
liyonghui160com
1. 环境描述
两个集群:rock 和 stone
rock无kerberos权限认证,stone有要求认证。
1. 从rock复制到stone,采用hdfs
Hadoop distcp -i hdfs://rock-nn:8020/user/cxz/input hdfs://stone-nn:8020/user/cxz/运行在rock端,即源端问题:报版本
- 一个备份MySQL数据库的简单Shell脚本
pda158
mysql脚本
主脚本(用于备份mysql数据库): 该Shell脚本可以自动备份
数据库。只要复制粘贴本脚本到文本编辑器中,输入数据库用户名、密码以及数据库名即可。我备份数据库使用的是mysqlump 命令。后面会对每行脚本命令进行说明。
1. 分别建立目录“backup”和“oldbackup” #mkdir /backup #mkdir /oldbackup
- 300个涵盖IT各方面的免费资源(中)——设计与编码篇
shoothao
IT资源图标库图片库色彩板字体
A. 免费的设计资源
Freebbble:来自于Dribbble的免费的高质量作品。
Dribbble:Dribbble上“免费”的搜索结果——这是巨大的宝藏。
Graphic Burger:每个像素点都做得很细的绝佳的设计资源。
Pixel Buddha:免费和优质资源的专业社区。
Premium Pixels:为那些有创意的人提供免费的素材。
- thrift总结 - 跨语言服务开发
uule
thrift
官网
官网JAVA例子
thrift入门介绍
IBM-Apache Thrift - 可伸缩的跨语言服务开发框架
Thrift入门及Java实例演示
thrift的使用介绍
RPC
POM:
<dependency>
<groupId>org.apache.thrift</groupId>
 是没有负载单原子时,载体的能量:
是没有负载单原子时,载体的能量: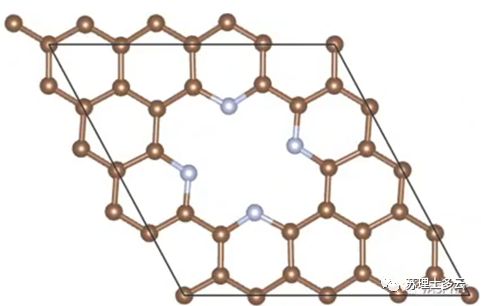 是每个单原子的能量,是用无限大的块体金属中平均每个金属的能量:
是每个单原子的能量,是用无限大的块体金属中平均每个金属的能量:
 答:因为我们用上述方法算出的结合能是和Bulk能量比较,所以一般算出来是正值,除了负载在石墨烯上的单原子具有较强的稳定性其结合能为负值;如果是和孤立的单原子比较的话其结合能就是负值了。
答:因为我们用上述方法算出的结合能是和Bulk能量比较,所以一般算出来是正值,除了负载在石墨烯上的单原子具有较强的稳定性其结合能为负值;如果是和孤立的单原子比较的话其结合能就是负值了。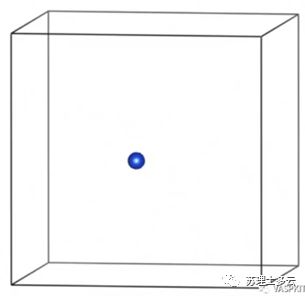 答:
答: