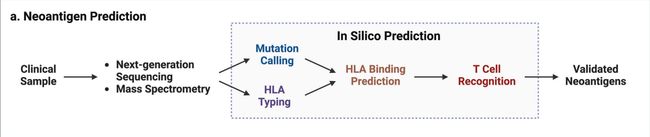
新抗原预测的计算工作流程
参考文献:Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. 2023 Jan 6;8(1):9. doi: 10.1038/s41392-022-01270-x. PMID: 36604431; PMCID: PMC9816309.
文章目录
-
-
- *新抗原预测的计算工作流程
-
- **Mutation calling:**
-
- NeuSomatic:
- INTEGRATE-neo(-):
- pVACtools:
-
- pVACseq
- pVACfuse:
- Epidisco(-):
- antigen.garnish:
- Spliceman2:
- Neopepsee:
- **HLA typing:**
-
- HLAreporter(-):
- PHLAT(-):
- *HLAscan:
- **HLA binding affinity:**
-
- pVACbind:
-
- *NetMHCpan:
- NeoPredPipe:
-
- ***how:**
- MixMHC2pred:
- neonmhc2:
- EDGE:
- **T cell recognition:**
-
- GLIPH2:
- *pMTnet:
-
*新抗原预测的计算工作流程
目前可用于体细胞突变新抗原预测的生物信息学流程有四个主要计算模块:(i)来自肿瘤WGS的HLA分型、WES数据和RNA-seq;(ii)使用一组体细胞突变和剪接变体的突变肽调用;(iii)HLA结合预测(HLA binding prediction);以及(iv)T细胞识别预测。

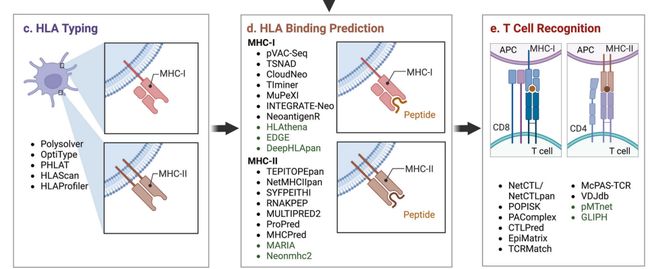
The in silico tools for mutation calling are listed as follows.
Mutation calling:
INTEGRATE-neo ,neoFusion,pVACtools,Epidisco,GATK, and Antigen.garnish,Spliceman,MutPred,REVEL,rMATS,pVACseq,Neopepsee,MuPeXI,RepeatMasker, CloudNeo,Tlminer, MuTect/MuTect2, Strelka/Strelka2,SMUFIN, VarScan2, SomaticSniper, CaVEMan, MuSE, cgpPindel, SvABA, RADIA, NeuSomatic, NeoantigenR, MutPred, JuncBase, Splice, SpliceGrapher, rMATS, SplAdder, ASGAL, REVEL, TSNAD,HERVd, HESAS and EnHERV,hervQuant.
NeuSomatic:
NeuSomatic基于深度卷积神经网络,用于精确的体细胞突变检测。有了经过适当训练的模型,它可以在测序平台、策略和条件下稳健地执行。NeuSomatic以一种新颖的方式总结和增强序列比对,并结合多维特征来有效地捕捉变异信号。它不仅是一种通用而且准确的体细胞突变检测方法。
bioinform/neusomatic: NeuSomatic: Deep convolutional neural networks for accurate somatic mutation detection (github.com)
Sahraeian, S.M.E., Liu, R., Lau, B. et al. Deep convolutional neural networks for accurate somatic mutation detection. Nat Commun 10, 1041 (2019). https://doi.org/10.1038/s41467-019-09027-x
INTEGRATE-neo(-):
ChrisMaherLab/INTEGRATE-Neo (github.com)
INTEGRATE Neo,用于使用NGS数据发现基因融合新抗原。INTEGRATE Neo扩展了高度精确的基因融合发现工具INTEGRATE的功能(Zhang et al.,2016)。将INTEGRATE-Neo应用于TCGA前列腺队列数据(PRAD),以证明其在识别可能作为个性化癌症免疫治疗靶点的基因融合新抗原方面的实用性。
基因融合(gene fusion):一文搞懂基因融合(gene fusion)的定义、产生机制及鉴定方法
pVACtools:
pVACtools是一个癌症免疫治疗工具套件,当与成熟的基因组学pipline配对时,可以为新抗原表征产生端到端的解决方案。pVACtools支持从不同机制鉴定改变的肽,包括点突变、帧内和移码插入和缺失以及基因融合。
pVACtools — pVACtools 4.0.1 documentation
Hundal, J. et al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 8, 409–420 (2020).
pVACtools — pVACtools 4.0.1 documentation
pVACtools是一个癌症免疫治疗工具套件,由以下工具组成:
pVACseq:癌症免疫治疗,用于从VCF文件中识别新抗原并确定其优先级。
pVACbind:癌症免疫治疗,用于从FASTA文件中识别新抗原并确定其优先级。
pVACfuse:一种用于检测基因融合产生的新抗原的工具。
pVACvector:一种专门用于帮助构建基于DNA的癌症疫苗的工具。
pVACview:基于R Shiny的应用程序,可帮助用户从pVACtools过程的结果中审查、探索新抗原并确定其优先级,以进行PGV设计。
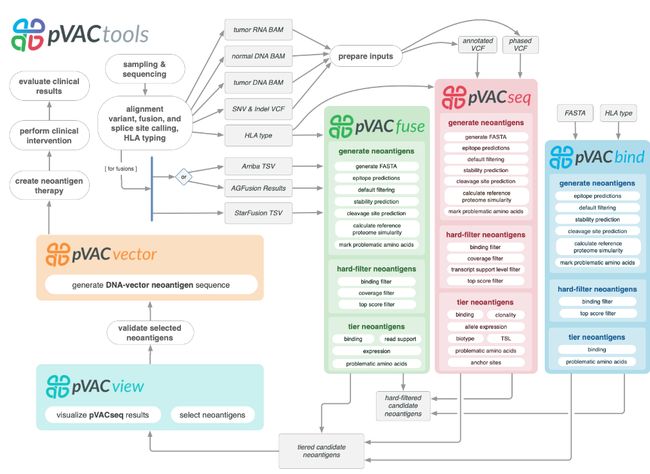
pVACseq
input:
用于从VCF文件中识别新抗原并确定其优先级。
.vcf文件:
(12条消息) 生信:1:vcf格式文件解读_vcf high_sor_genome_denovo的博客-CSDN博客
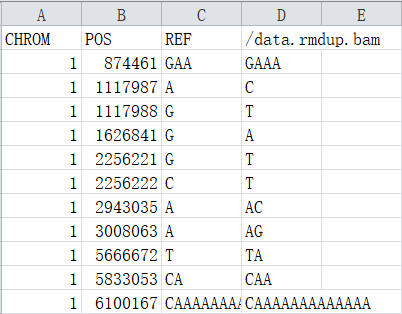
VCF文件分为两部分内容:以“#”开头的注释部分和没有“#”开头的主体部分。(注释部分和主题部分);注释部分有很多对VCF的介绍信息;主体部分包含10列数据。主题部分每一行代表一个variant的信息。
注释:

主体部分 :

CHROM : 参考序列名称
POS : variant所在的left-most位置(1-base position)(发生变异的位置的第一个碱基所在的位置)
生信格式中的0-base与1-base - 简书 (jianshu.com)

ID : variant的ID。同时对应着dbSNP数据库中的ID,若没有,则默认使用‘.’
REF : 参考序列的Allele,(等位碱基,即参考序列该位置的碱基类型及碱基数量)
ALT : variant的Allele,若有多个,则使用逗号分隔,(变异所支持的碱基类型及碱基数量)这里的碱基类型和碱基数量,对于SNP来说是单个碱基类型的编号,而对于Indel来说是指碱基个数的添加或缺失,以及碱基类型的变化
人类基因组上的变异主要分为三大类:
1. 单核苷酸变异,(通常称为单核苷酸多态性,通俗的说法就是单个DNA碱基的不同,简称SNP);
2. 小的Indel(Insertion 和 Deletion的简),指的是在基因组的某个位置上所发生的小片段序列的插入或者删除,其长度通常在50bp以下(这个长度范围的变异可以利用Smith-Waterman 的比对算法来获得1,2);
3. 大的结构性变异,这种类型比较多,包括长度在50bp以上的长片段序列的插入或者删除、染色体倒位,染色体内部或染色体之间的序列易位,拷贝数变异,以及一些形式更为复杂的变异。为了和SNP变异作区分,第2和第3类变异通常也被称为基因组结构性变异(Structural variation,简称SV)。
QUAL : variants的质量。Phred格式的数值,代表着此位点是纯合的概率,此值越大,则概率越低,代表着次位点是variants的可能性越大。(表示变异碱基的可能性)
FILTER : 次位点是否要被过滤掉。如果是PASS,则表示此位点可以考虑为variant。
基因:是DNA分子中的一段恰好能表达一种蛋白质的序列, 可以连续也可以不连续,但是在转录过程中是联动的。以前的说法是,-个基因一个性状(character),但是性状的定义太模糊了。实际运用中,就是一个基因一种蛋白质。
染色体:细胞有丝分裂前期,细胞核内的DNA分子在组蛋白的引导下缠绕折叠,在显微镜下形成X形或Y形的易于被碱性燃料染色的结构。
位点:就是一对碱基在基因片段里的位置,相当于一列队伍里的序号。
SNP位点:单核苷酸多态性位点,在一个能正常表达蛋白质的基因序列中,有些位置.上的核苷酸不-定严格的是ACGT当中的一-种,可以是两种、三种、或者全部。也就是说,把这个位置上的碱基替换成其他的,这个基因的功能还是正常的。这个位点叫SNP位点。
INFO : variant的相关信息
FORMAT : variants的格式,例如GT:AD:DP:GQ:PL
SAMPLES : 各个Sample的值,由BAM文件中的@RG下的SM标签所决定,这些值对应着第9列的各个格式,不同格式的值用冒号分开,每一个sample对应着1列;多个samples则对应着多列,这种情况下列的数多余10列。

从VCF文件提取出与基因组不一致的SNP位点
output:
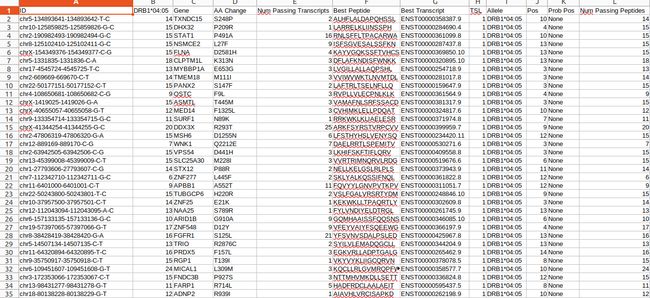
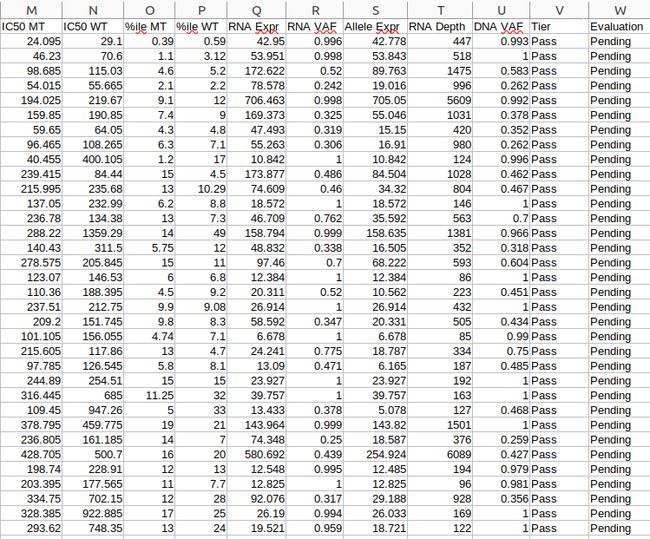
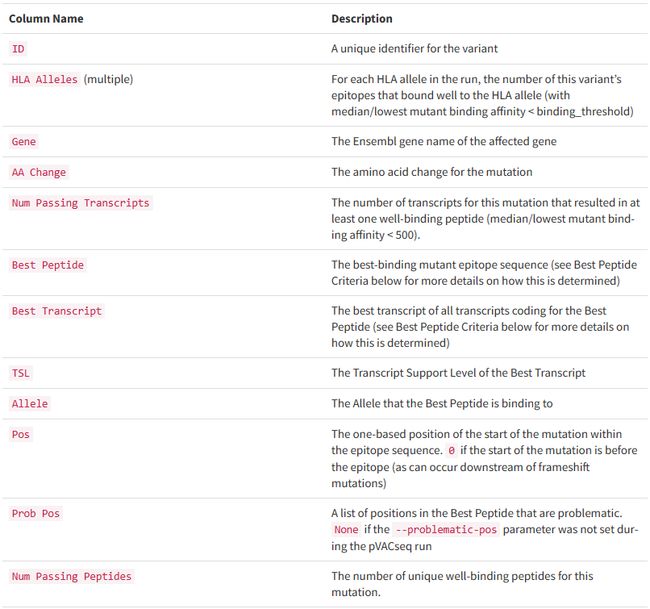
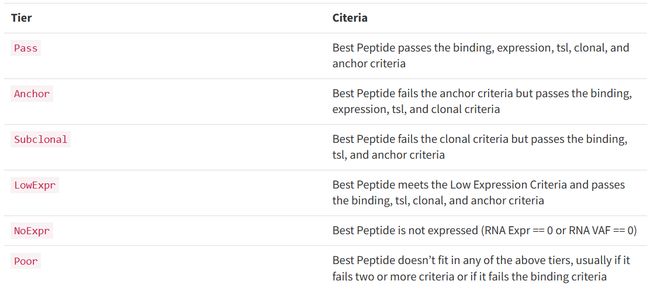
all_epitopes.aggregate.tsv文件是all_epitopes tsv的聚合版本。它显示了每个变体的最佳评分表位,并输出该表位的额外结合亲和力、表达和覆盖率信息。它还提供了每个变体的良好评分表位总数、这些表位覆盖的转录物数量,以及这些表位与之结合良好的HLA等位基因的信息。最后,该报告将变体分为几层,为变体在疫苗中的适用性提供建议。只有符合聚集包含阈值的表位才会包含在此报告中(默认值:5000)。在IC50 MT、IC50 WT、%ile MT和%ile WT列中输出的中值或最低结合亲和力度量由-最高得分度量参数控制。
Output Files — pVACtools 4.0.1 documentation

*how?

pVACfuse:
input:
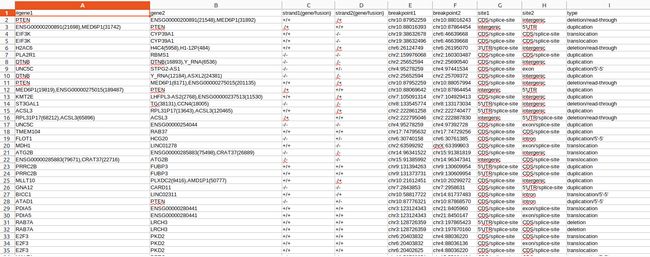
output:


Epidisco(-):
Epidisco用于建立本地或集群计算环境,安装所有相关的生物信息学工具,并协调这些不同工具对输入数据的执行。
Epidisco在两个层面上加速了基因组学管道的部分。独立的计算任务,如QC检查、RNA测序数据的处理和正常肿瘤DNA测序数据的联合分析,都是并行运行的。在每个工具的调用中,如果可能,将测序数据拆分为多个片段,并行计算部分结果,然后合并。
hammerlab/epidisco: Personalized cancer epitope discovery and peptide vaccine prediction pipeline (github.com)
协调PGV(personalized genomic vaccine) pipeline执行的工作流管理工具称为Epidisco
Epidisco用于建立本地或集群计算环境,安装所有相关的生物信息学工具,并协调这些不同工具对输入数据的执行。
Epidisco在两个层面上加速了基因组学管道的部分。独立的计算任务,如QC检查、RNA测序数据的处理和正常肿瘤DNA测序数据的联合分析,都是并行运行的。在每个工具的调用中,如果可能,将测序数据拆分为多个片段,并行计算部分结果,然后合并。
Computational Pipeline for the PGV-001 Neoantigen Vaccine Trial
personalized genomic vaccine (PGV)
PGV-001是一种针对从患者肿瘤样本中鉴定的新抗原的治疗性肽疫苗。肽是通过计算管道选择的,该管道从肿瘤/正常外显子组测序中识别突变,并通过预测的I类MHC亲和力和从肿瘤RNA中估计的丰度的组合对突变序列进行排序。个性化基因组疫苗(PGV)管道是模块化的,由独立可用的工具和软件库组成。
协调PGV管道执行的工作流管理工具称为Epidisco(cite: Mondet S, Aksoy BA, Rozenberg L, Hodes I, Hammerbacher J. Bioinformatics Workflow Management With The Wobidisco Ecosystem. bioRxiv; 2017. DOI: 10.1101/213884.)。Epidisco用于建立本地或集群计算环境,安装所有相关的生物信息学工具(外部软件,如GATK,以及我们自己的工具,包括Isovar和Vaxrank),并协调这些不同工具对输入数据的执行。
Epidisco在两个层面上加速了基因组学管道的部分。独立的计算任务,如QC检查、RNA测序数据的处理和正常肿瘤DNA测序数据的联合分析,都是并行运行的。在每个工具的调用中,如果可能,将测序数据拆分为多个片段,并行计算部分结果,然后合并。
Epidisco支持本地计算、传统的HPC调度程序(如LSF)以及来自谷歌云和AWS的基于云的资源。在一台典型的机器上,为一名患者运行完整的PGV管道可能需要长达4天的时间;但是,使用五台或五台以上的计算机进行并行化可以将总体运行时间减少到一天
Epidisco还使PGV管道能够容忍中间步骤的故障,并允许通过简单的重新启动请求从故障点恢复管道。通过以自动化的方式处理此类故障,执行清洁程序,只重新启动需要重新运行的任务,该工作流程使研究人员更容易操作此类复杂的计算任务。Epidisco提供了命令行和基于web的实用程序,以方便启动新的工作流程、收集结果以及对管道的特定部分进行故障排除。
PGV管道使用的各个基础设施工具被实现为OCaml堆栈,包括:

antigen.garnish:
antigen.garnish是一个开源的肿瘤抗原质量分析R包。318例癌症样本中,antigen.garnish预测了291,376个主要组织相容性复合体(MHC)结合推测的新抗原。通过对318例癌症患者的5个临床数据集进行分析发现:在患者的肿瘤中,差异性大的肿瘤抗原具有特异性,其富含疏水序列,并与非小细胞肺癌患者在PD-1治疗后的生存率相关。
该模型研究的是来自患者个体样本的蛋白质序列,评估其中有多少与健康细胞相似,又有多少与健康细胞不同(这些不同足以使免疫系统产生反应)。它越不同,它的免疫治疗目标就越好,因为它更有可能激活治疗,对健康细胞的附带损害越少。该模型的预测也个性化到每个病人的样本。
SNV和复杂变体对人和小鼠肿瘤新抗原的预测。
andrewrech/antigen.garnish (github.com)
Richman LP, Vonderheide RH, and Rech AJ. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Systems. 2019.
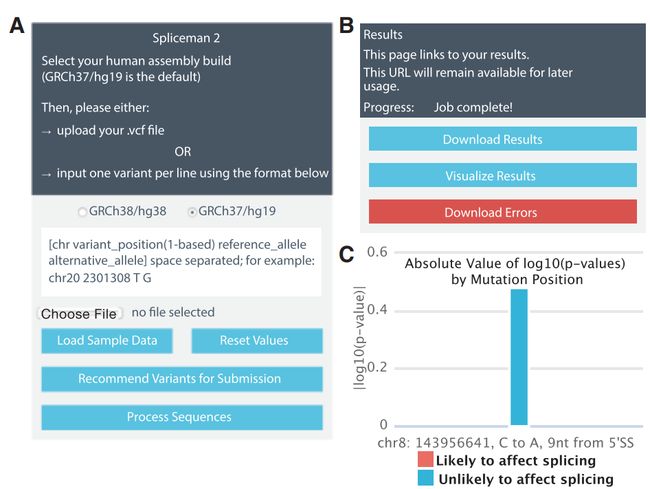
Spliceman2:
mehods: Fairbrother Lab Website (brown.edu)
Spliceman2 :Fairbrother Lab Website (brown.edu)

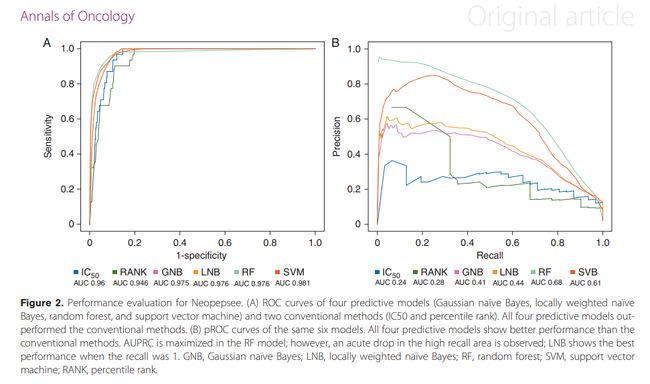
Neopepsee:
一种用于下一代测序数据的基于机器学习的新抗原预测程序。利用原始RNA-seq数据和体细胞突变列表,Neopepsee自动提取突变肽序列和基因表达水平。测试了14个免疫原性特征来构建机器学习分类器,并与基于IC50的传统方法在敏感性和特异性方面进行了比较。在黑色素瘤、白血病和癌症的独立数据集上测试了Neopesee。
Neopepsee / Wiki / Home (sourceforge.net)
Kim S, Kim HS, Kim E, Lee MG, Shin EC, Paik S, Kim S. Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann Oncol. 2018 Apr 1;29(4):1030-1036. doi: 10.1093/annonc/mdy022. PMID: 29360924.
Neopepsee download | SourceForge.net
HLA typing:
Polysolver,OptiType,HLAreporter, PHLAT,HLAScan,HLAProfiler.
HLAreporter(-):
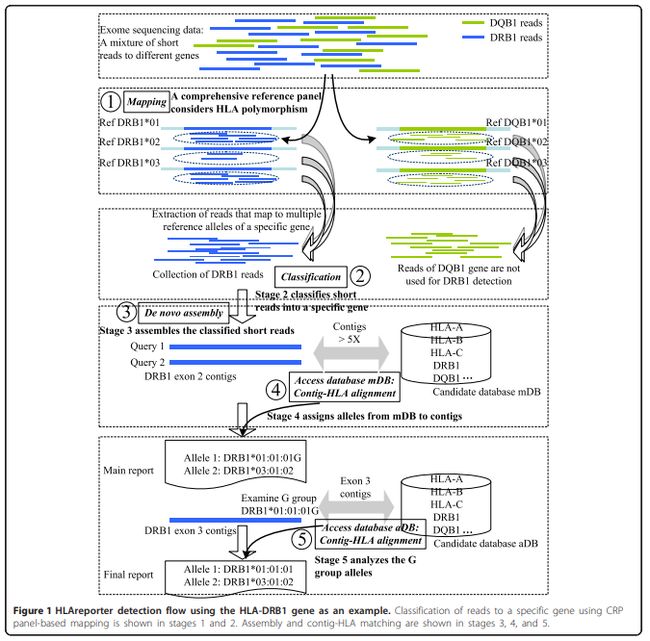
用于从NGS数据中进行HLA分型,该工具基于读取映射,使用包含所有已知HLA等位基因的单倍型参考序列集(Reference panel),**然后重新组装基因特异性短读取。**在公开的NGS数据上进行测试时,实现了高数字分辨率的精确HLA分型,优于其他新开发的工具,如HLAminer和PHLAT。
PHLAT(-):
PHLAT是一种计算工具,旨在使用RNAseq或外显子组测序数据作为输入,对主要的I类和II类HLA基因进行高分辨率(4位数)分型。
PHLAT结果解读
进入results目录下,查看example_HLA.sum文件。
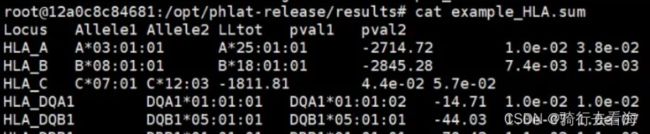
这个结果各个列代表的含义:
Locus表示基因座,人类 6 号染色体上有六个 HLA 基因座:HLA-A、HLA-B、HLA-C、HLA-DQA1、HLA-DQB1 和HLA-DRB1。每个基因座都有一对等位基因,在“Allele1”和“Alllele2”列中定义。
Allele1: 第一个等位,这里要注意的是HLA基因座与它的分辨率之间用“*”分隔。PHLAT工具分辨率最低是4位。如果“Allele1”和“Alllele2”中的条目相同,则该基因座是纯合的。如果它们不同,则该基因座是杂合的。如果报告了多个预测,该等位基因可能被视为不明确的。
**Allele2:**另一个等位。
**LLtot:**报告的配对等位基因对(Allele1, Allele2)的对数似然得分。
pval1: Allele1的p值。
pval2: Allele2的p值。
*HLAscan:
HLAscan对来自国际ImMunoGeneTics项目/人类白细胞抗原**(IMGT/HLA)数据库的HLA序列进行比对**。比对读数的分布用于计算得分函数,以通过逐步去除假阳性等位基因来确定正确的等位基因。
HLAscan是一个基于比对的程序,它在考虑阅读分布的情况下确定单倍型。该软件对国际ImMunoGeneTics项目/人类白细胞抗原(IMGT/HLA)数据库中的HLA序列进行比对。
比对读数的分布用于计算得分函数,以通过逐步去除假阳性等位基因来确定正确的等位基因。HLAscan可以可靠地应用于全基因组、外显子组和靶序列的HLA类型测定。
SyntekabioTools/HLAscan (github.com)
HLAScan是由韩国的科研团队开发的一款HLA分型工具,可以处理WGS, WES和目标区域捕获测序的数据,将reads与IMGT/HLA数据库中的reads进行比对,然后确定HLA的基因型信息。
软件的开发团队利用测试数据,评估了以下几个软件的分型效果。
可以看到HLAscan能够提供可靠的2位和4位分型的结果。
input:
FASTQ
从计算机的角度来说,生物的序列属于一种字符串,也是一种文本,因此生物信息分析属于文本处理范畴。文本存储为固定格式文件,生物信息的工作就是各种文本文件之间格式的转换,例如通过序列拼接将fastq转换为fasta,通过短序列比对将fastq与fasta合并为bam,通过变异检测将bam中突变位点提取出来转换为vcf。因此,我们可以通过总结每一种生物数据文件格式的处理方法来学习生物信息,这样当拿到固定格式的文件之后,就知道该如何来处理了。
生信数据格式说明 - FASTQ文件 - 知乎 (zhihu.com)
Fastq格式是一种基于文本的存储生物序列和对应碱基(或氨基酸)质量的文件格式。
下面是一个Illumina平台测序的真实数据,其中包含了1条reads的信息。
@ST-E00126:128:HJFLHCCXX:2:1101:7405:1133
TTGCAAAAAATTTCTCTCATTCTGTAGGTTGCCTGTTCACTCTGATGATAGTTTGTTTTGG
+
FFKKKFKKFKFFASTQ格式储存的序列信息,每1条reads的信息,可以分成4行。
第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;
@ST-E00126:128:HJFLHCCXX:2:1101:7405:1133
@ 开始的标记符号
ST-E00126:128:HJFLHCCXX 测序仪唯一的设备名称
2 lane的编号
1101 tail的坐标
7405 在tail中的X坐标
1133 在tail中的Y坐标
第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。
sam和bam文件
(13条消息) 生信分析过程中这些常见文件(fastq/bed/gtf/sam/bam/wig)的格式以及查看方式你都知道吗?_生信宝典的博客-CSDN博客
sam文件全称The Sequencing Alignment/Map Format,用于存储reads比对到参考基因组的比对结果,是一个纯文本格式,文件一般较大。为了节省硬盘存储,一般使用其高效压缩的二进制格式bam文件。
利用samtools view的-b参数就能把sam文件转为bam文件。
1)sam文件查看方式
在linux终端直接用less即可进行查看;
2)bam文件查看方式
需要借助samtools view工具进行查看
samtools view filename.bam | less -S
samtools view -h filename.bam | less -S
NGS分析中大多数文件都是由header和record两部分组成,加上-h参数后可以将header显示出来,默认是不显示的。
@HD VN:1.5 SO:coordinate
@SQ SN:chr1 LN:248956422
@SQ SN:chr10 LN:133797422
......
@SQ SN:chrKI270392.1 LN:971
@SQ SN:chrKI270394.1 LN:970
@RG ID:BH_H3K27ac_2 LB:BH_H3K27ac_2 SM:BH_H3K27ac_2
@PG ID:bwa PN:bwa VN:0.7.15-r1140 CL:bwa mem -M -t 8 -R @RG\tID:BH_H3K27ac_2\tLB:BH_H3K27ac_2\tSM:BH_H3K27ac_2\tPL: /MP
@PG ID:MarkDuplicates VN:1.138(aa51703435dc6a423013e74e56b0b68405facd79_1439324166) CL:picard.sam.markduplicates.
K00141:244:HVL3NBBXX:8:2119:27235:31453 99 chr1 10016 32 115M = 10016 115 CCCTAACCCTAACCCTAACCC
K00141:244:HVL3NBBXX:8:2119:27235:31453 147 chr1 10016 32 115M = 10016 -115
CCCTTACCCTAACCCTAACCC
header内容
@HD:是必须的标准文件头,包含版本信息;
@SQ:参考序列染色体名字和长度信息 (SN: scaffold name; LN: length);
@RG:重要read group信息,通常包含测序平台,测序文库和样本ID等信息,分析时用于区分不同样本(重测序时用到);
@PG:生成此文件的操作过程和参数信息 (program)。
record内容
每一行就是一条read比对上参考基因组的信息,总共12列,用tab键分割。
# 1. read名称;
# 2. 比对信息位flag值;
# 3. 参考序列染色体编号;
# 4. 5′端起始位置;
# 5. MAPQ:mapping quality,描述比对的质量,数字越大,特异性越高;
# 6. CIGAR字符串,记录插入、删除、错配等信息;
# 7. 配对read所比对到的染色体,仅双端测序的数据才有;
# 8. 配对read所比对到的位置,仅双端测序的数据才有;
# 9. 插入片段的长度,仅双端测序的数据才有;
# 10. read序列;
# 11. read质量值;
# 12. 12列以后的信息都是metadata,程序用标记
output:
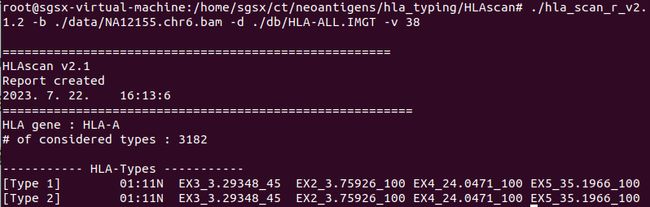
可以看到,对于HLA-A基因,基因分型的结果为HLA-A*01:11N。

how:

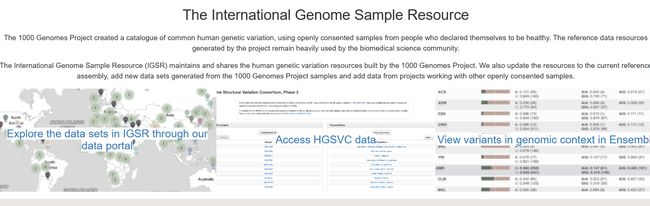
1000 Genomes | A Deep Catalog of Human Genetic Variation (internationalgenome.org)
HLA binding affinity:
NetMHCpan,NetMHCIIpan4.0,MixMHC2pred,MARIA,neomhc2,pVAC-Seq, TIminer, HLAthena, DeepHLApan, TEPITOPEpan, NetMHCIIpan, SYFPEITHI, RNAKPEP, MULTIPRED2, ProPred, MHCPred, MARIA, Neonmhc2, EDGE.
pVACbind:
pVACbind:癌症免疫治疗,用于从FASTA文件中识别新抗原并确定其优先级。
Hundal, J. et al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 8, 409–420 (2020).
imput:
pVACbind uses a FASTA file as its input.

FASTA 格式文件中的每个序列信息由两个部分组成:
1. 描述行 (The description line, Defline, Header or Identifier line): 以一个大于号(“>”)开头,内容可以随意,但不能有重复,相当于身份识别信息。
2. 序列行 (Sequence Line):一行或多行的核苷酸序列或肽序列,其中碱基对或氨基酸使用单字母代码表示。
特点:简单,易于理解
output:
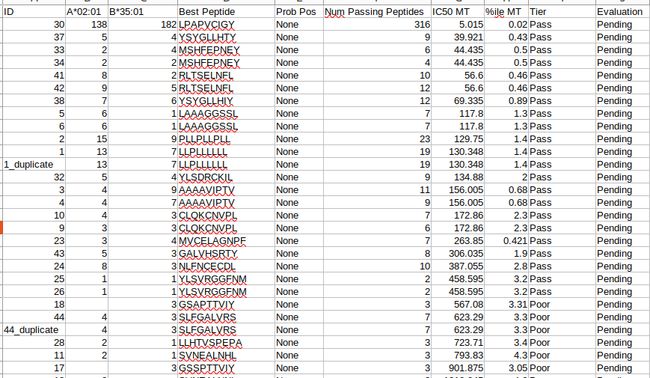

*NetMHCpan:
NetMHCpan软件用于预测肽段与MHC I型分子的亲和性,最新版本为v4.0, 基于人工神经网络算法,以180000多个定量结合数据和MS衍生的MHC洗脱配体的组合为训练集构建模型。结合亲和力数据来自人,小鼠,猪等多个物种的MHC分子,MS洗脱的配体数据来自55个人和小鼠的HLA等位基因。
NetMHCpan 4.1 - DTU Health Tech - Bioinformatic Services

第一步上传涵盖了体细胞突变位点的氨基酸序列,上传的氨基酸序列是突变之后的序列,不是野生型的蛋白质序列。
第二步选择切割肽段的方式,抗原通过抗原表位与MHC分子结合,MHC I型分子可以结合的抗原表位长度为8到11个氨基酸,对应这里的8-11mer,先将蛋白质序列切分成短的肽段之后在进行MHC分子亲和性的预测。
第三步选择HLA allel, 确定之后点击提交按钮即可。输出结果示意如下
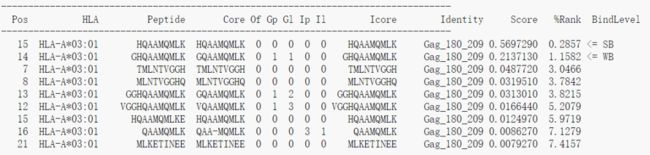
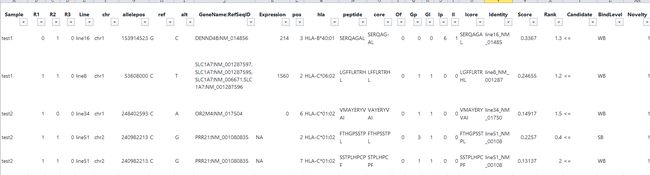
列数很多,其中的Peptide就是从原始的输入序列中提取出的长度为8-11个氨基酸的肽段,Pos对应肽段的在原始序列上的起始位置,第一个位置从0开始计数;Core对应与MHC结合的肽段序列,和blast类似,允许插入和缺失,%Rank代表该肽段是一个天然存在的肽段的可能性,数值越小越好,最后一列的BindLevel代表亲和力的强弱水平,SB表示strong binding, WB表示weak bingding。每一列的详细解释参见以下链接
http://www.cbs.dtu.dk/services/NetMHCpan/output.php
NeoPredPipe:
数据分析pipeline,只需要提供肿瘤患者的体细胞突变数据和HLA分型结果即可,软件自动提取突变氨基酸序列,并进行NetMHCpan分析
https://github.com/MathOnco/NeoPredPipe
input:
-I指定体细胞突变的vcf文件,-H指定HLA分型结果文
1、VCF file(s)

2、hla file
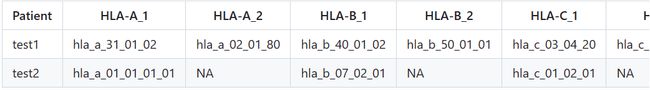
output:
*how:
Łuksza, M., Riaz, N., Makarov, V. et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520 (2017).
MixMHC2pred:
MixMHC2pred是显示在细胞表面的HLA-II配体的预测因子。它结合了对MHC-II分子亲和力的预测以及与抗原处理和呈递相关的其他特征。它可以对任何人类MHC-II等位基因(HLA-DR/-DP/-DQ)以及其他物种的许多等位基因起作用。
GfellerLab/MixMHC2pred: HLA-II ligand predictor. (github.com)
MixMHC2pred Server (gfellerlab.org)

neonmhc2:
neonmhc2.org
EDGE:
将深度学习应用于来自各种人类肿瘤的大型(N=74名患者)HLA肽和基因组数据集,以创建用于新抗原预测的抗原呈递计算模型。HLA抗原预测的阳性预测值提高了九倍。将EDGE应用于使用常规临床标本和少量合成肽鉴定最常见HLA等位基因的新抗原和新抗原反应性T细胞。EDGE可以提高癌症患者开发新抗原靶向免疫治疗的能力。
T cell recognition:
NetCTL/NetCTLpan, POPISK, PAComplex, CTLPred, EpiMatrix, TCRMatch
GLIPH2:
GLIPH算法,该算法基于互补决定区3(CDR3)中的共有相似性对识别相同表位的TCR进行聚类,并根据受试者的基因型预测它们的HLA限制性。在本次研究中,研究人员在算法优化后,开发了GLIPH2的改进算法,这种算法可以快速分析数百万个TCR序列,聚类效率和准确率都得到了提高
GLIPH 2
CD4+T细胞对对抗病原体至关重要,但HLA等位基因的多样性(>20000)和许多病原体基因组的复杂性阻碍了对人类T细胞特异性的全面分析。我们之前描述了GLIPH,这是一种对识别相同表位的T细胞受体(TCRs)进行聚类并预测其HLA限制性的算法,但当分析>10000个TCRs时,这种方法失去了效率和准确性。在这里,我们描述了一种改进的算法GLIPH2,它可以处理数百万个TCR序列。我们使用GLIPH2分析了来自58名潜伏感染结核分枝杆菌(Mtb)的个体的19044个独特的TCRβ序列,并根据其特异性对其进行分组。为了鉴定Mtb特异性T细胞簇靶向的表位,我们使用人工抗原呈递细胞(aAPC)和报告T细胞对3724种不同的蛋白质进行了筛选,覆盖了95%的Mtb蛋白质编码基因。我们发现至少有五种PPE(Pro-Pro-Glu)蛋白是Mtb中T细胞识别的靶标。
通常,用户将传入一组数百到数千个TCR序列。该数据集将针对非常相似的TCR或共享CDR3基序的TCR进行分析,相对于未选择的原始参考TCR集中的预期频率,这些基序在该集合中似乎富集。
GLIPH返回重要的基序列表、重要的TCR收敛组,并且对于每个组,该组的分数集合指示基序、V基因、CDR3长度、贡献者之间共享的HLA和增殖计数的富集。当HLA数据可用时,它预测TCR集合识别的可能HLA。

*pMTnet:
pMHC-TCR结合预测网络(pMTnet),该模型基于迁移学习,用来预测新生抗原(neoantigens)和T细胞受体(T-cell receptors, TCRs)结合特异性。模型仅需给定TCR序列和(新生)抗原序列以及MHC类型即可完成TCR结合特异性预测,并进行一系列基因组范围的分析
tianshilu/pMTnet: Deep Learning the T Cell Receptor Binding Specificity of Neoantigen (github.com)
Code availability The pMTnet software is available on GitHub at https://github.com/tianshilu/pMTnet73. Pipeline for HERV expression detection is available on GitHub at https://github.com/jcao89757/HERVranger74. QBRC mutation calling pipeline is available on GitHub at https://github.com/tianshilu/QBRC-Somatic-Pipeline75. QBRC neoantigen calling pipeline is available on GitHub at https:// github.com/tianshilu/QBRC-Neoantigen-Pipeline76
input:
-
input.csv:input.csv文件,包含3列,命名为“CDR3,抗原,HLA”:TCRβCDR3序列、肽序列和HLA等位基因。
-
- T细胞介导的抗原识别取决于T细胞受体(TCR)与抗原主要组织相容性复合体(MHC)分子的相互作用 由于CDR3是与抗原直接接触的TCR区域,因此CDR3在TCR与肽-MHC复合物的相互作用中起到了十分重要的作用。 For more details about CDR3 encoding, please refer to https://github.com/jcao89757/TESSA.
For more details about CDR3 encoding, please refer to https://github.com/jcao89757/TESSA.
output:
pMTnet输出一个包含4列的表:CDR3序列、抗原序列、HLA等位基因,以及每对TCR/pMHC的排名。该等级反映了TCR和pMHC之间的预测结合强度相对于10000个随机采样的TCR对相同pMHC的百分位数等级。较低的级别被认为是一个好的预测。10000个背景TCR的序列可以折叠

how:

transfer learning-based model

**TCR-抗原结合特异性。**为降低预测任务的难度,作者将此分为3个步骤。首先,作者训练一个LSTM网络(长短期记忆(Long short-term memory, LSTM))(图1c),来获得抗原和MHCs的蛋白序列的嵌入(仅Ⅰ类)。其次,作者利用堆叠的自编码器训练TCR序列嵌入(图1a),再次编码TCR序列。最后,在这些嵌入外创造一个深度神经网络,以将来自TCR、抗原肽序列和MHC等位基因的知识以具有生物学意义的方式结合。(图1e)。作者还通过微调确定TCRs和pMHCs预测模型。作者创建了10倍于正例数量的负例,训练150轮结果如图1f所示。
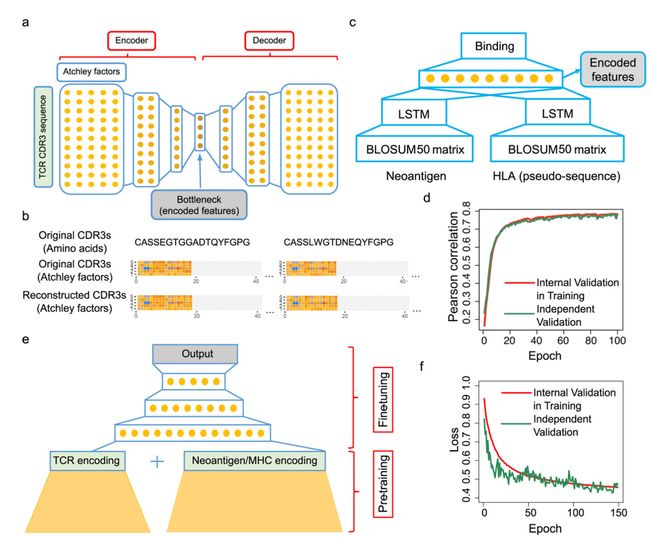
本文模型仅需给定TCR序列和(新生)抗原序列以及MHC类型即可完成TCR结合特异性预测,并进行了一系列基因组范围的分析。该模型还可用于治病治疗以及设计TCR-T或新生抗原治疗。总体而言,本文证明了TCR和PMHCs之间的配对,仅给予TCR、抗原和MHC序列的情况下是机器可学习的,这为将来产生更高精度TCR抗原预测模型的研究奠定了基础。